
代谢工程改造大肠杆菌生产氢咕啉酸
2023-10-18 00:26董宁房欢赵莹姜平涛赵江华张同存张大伟
食品与发酵工业 2023年19期
董宁,房欢,赵莹,姜平涛,赵江华,4,张同存,张大伟*
1(天津科技大学 生物工程学院,天津,300457)2(中国科学院天津工业生物技术研究所,低碳合成工程生物学重点实验室,天津,300308)3(国家合成生物技术创新中心,天津,300308)4(中国科学院大学,北京,300192)5(工业发酵微生物教育部重点实验室,天津市工业微生物重点实验室,天津科技大学生物工程学院,天津,300457)
维生素B12,又名钴胺素,是唯一含有金属元素的维生素,也是结构最复杂的维生素。其主要包括氰钴胺素、腺苷钴胺素、甲钴胺素和羟钴胺素四类,其中腺苷钴胺素和甲钴胺素是生物体中具有活性的分子[1-2]。维生素B12只能通过细菌和古细菌生成,人体不能合成[3]。它是微生物代谢以及人类生长发育所必须的营养成分。工业生产维生素B12主要利用脱氮假单胞菌(Pseudomonasdenitrifican)、弗氏丙酸杆菌(Propionibacteriumfreudennreichii)和苜蓿中华根瘤菌(Sinorhizobiummeliloti)等微生物发酵[4-6]。而近几年通过合成生物学和代谢工程技术构建的大肠杆菌已经可以用来生产维生素B12[7-8]。维生素B12作为生物体中DNA、氨基酸代谢等重要的辅酶,已被广泛应用于医药和食品行业。
氢咕啉酸(hydrogenobyrinic acid,HBA)外观呈橙红色,是维生素B12合成途径中比较稳定的中间体[9],合成途径如图1所示。提高HBA的产量对维生素B12产量提高有至关重要的作用。我们前期构建了合成HBA的大肠杆菌工程菌[7-9],虽然产量较高,但是工程菌具有抗性,不利于生产。选择稳定、表现良好的宿主细胞对于大规模生产目的分子至关重要[10]。辅因子和前体途径优化、减少副产物合成是构建工业微生物生产菌株的常用策略[11-13]。
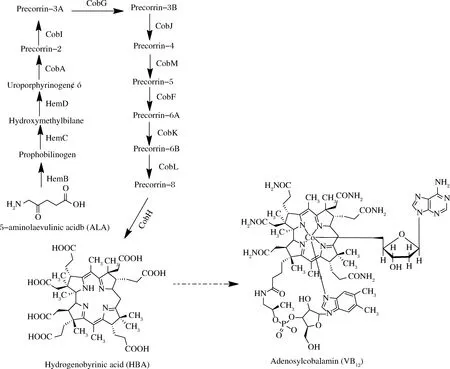
图1 维生素B12生物合成途径Fig.1 Vitamin B12 biosynthetic pathway
本研究选取了6种不同的大肠杆菌作为宿主,分别是BL21(DE3)、BL21 Star(DE3)、MG1655(DE3)、BW25113(DE3)、JM109(DE3)和shuffie T7-K12。将HBA合成质粒分别转化到上述6种大肠杆菌宿主细胞中,发酵并进行HPLC检测,发现BW25113(DE3)是最佳宿主。为了缓解质粒表达对菌株所带来的代谢负担,采取一系列代谢工程策略,如将HBA模块整合到BW25113(DE3)基因组的hsdR基因位点,引入合成前体尿卟啉原Ⅲ模块,敲除代谢副产物乙酸合成基因ackA-pta,引入苜蓿中华根瘤菌和运动发酵假单胞菌(Zymomonasmobilis)两种来源的Entner-Doudoroff(ED)途径提高还原型烟酰胺腺嘌呤二核苷酸磷酸(nicotinamide adenine dinucleotide phosphate,NADPH)供给,得到了无抗生产HBA高产菌种,为下一步高产维生素B12工程菌的构建奠定了基础。
1 材料与方法
1.1 材料
1.1.1 实验菌株与质粒
论文中涉及的菌株见表1,质粒见表2。

表1 本实验所用菌株Table 1 Strains used in this study
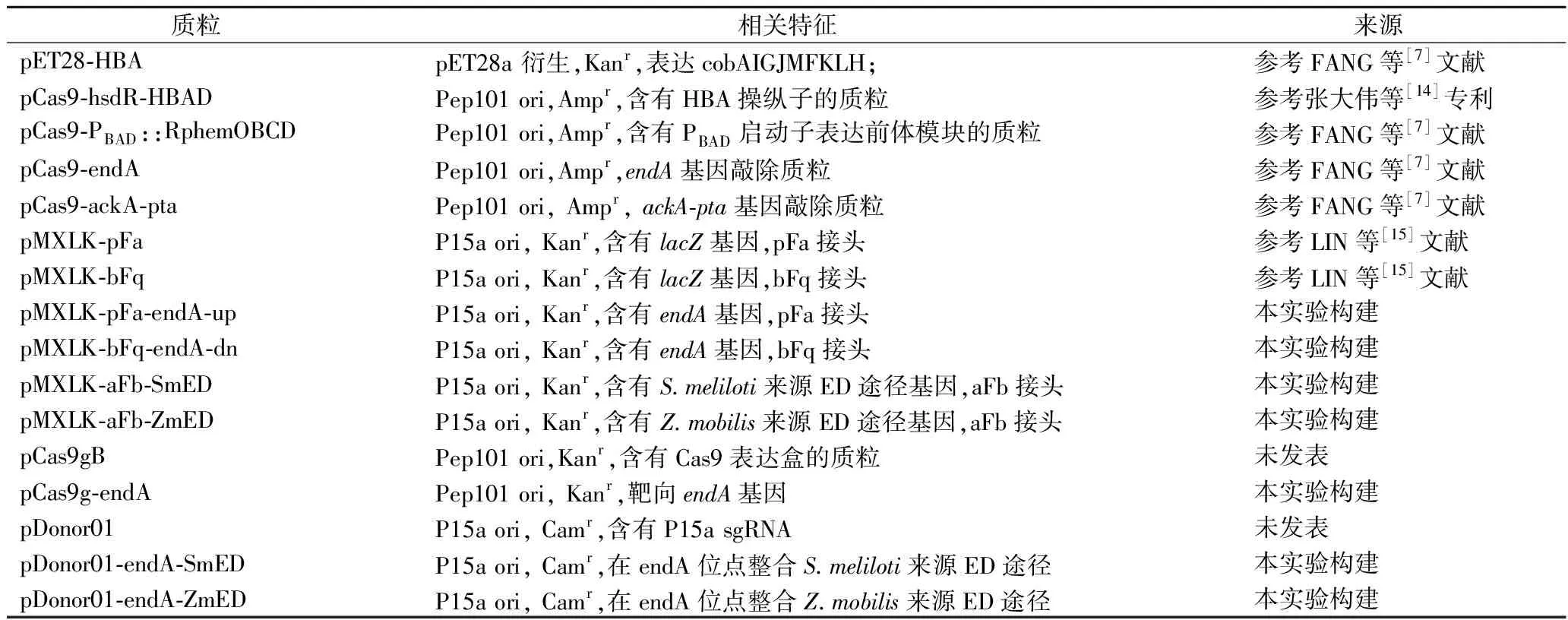
表2 本实验所用质粒Table 2 Plasmids used in this study
1.1.2 引物
本实验所涉及的基因序列均来自NCBI(https://www.ncbi.nlm.nih.gov/),使用Primer Premier 6或SnapGene设计所用引物,由北京擎科生物公司合成(表3)。
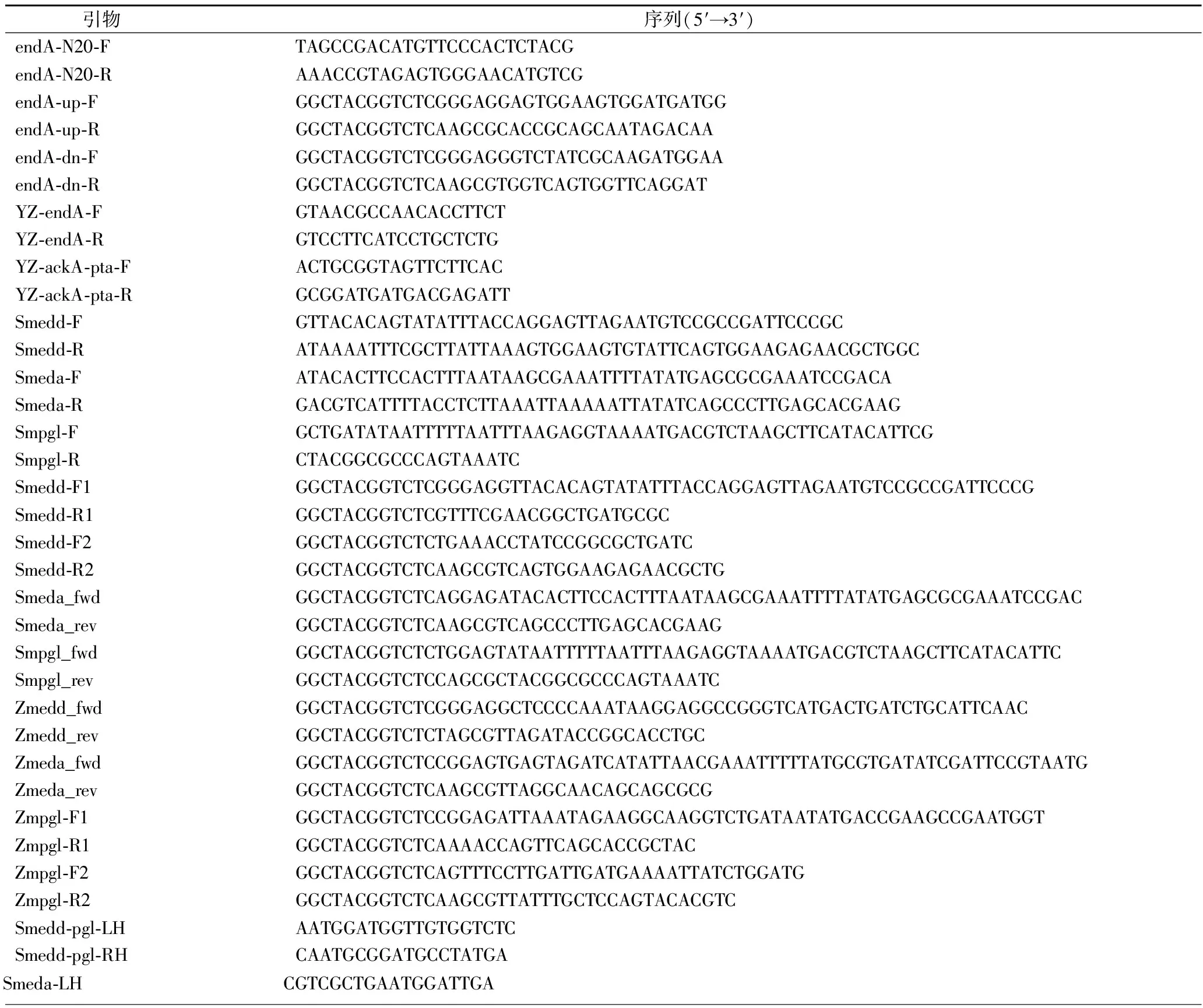
表3 本实验所用引物Table 3 Primers used in this study
1.1.3 主要培养基
LB(Luria-Bertani)液体培养基(g/L):蛋白胨10,酵母抽提物5,NaCl 10,pH 7.5,121 ℃灭菌20 min。
LB固体培养基:在调好pH值的LB液体培养基加入15 g/L的琼脂粉,121 ℃灭菌20 min。
TYGly培养基(g/L):酵母提取物5、胰蛋白胨10、KH2PO45、甘氨酸2、甘油10、琥珀酸10、甜菜碱15,用NaOH调节初始pH值至7.0~7.2,121 ℃灭菌20 min。
培养基在接种前按需添加相应抗生素,其中氨苄青霉素(ampicillin,Amp)质量浓度为100 μg/mL,硫酸卡那霉素(kanamycin sulfate,Km)质量浓度为50 μg/mL,氯霉素(chloramphenicol,Cm)质量浓度为34 μg/mL。
1.1.4 仪器与设备
TC-XP-G PCR仪,BioER;EYETMII凝胶自动成像仪,法国VILBER LOURMAT;V-1600可见分光光度计,上海美谱达仪器有限公司;DYY-6c电泳仪,北京市六一仪器厂;HPX-9162 MBE电热恒温培养箱,上海博讯实业有限公司医疗设备厂;ND 2000 NanoDrop超微量分光光度计,Thermo Fisher;LDZX-50 KB立式压力蒸汽灭菌锅,上海申安医疗器械厂。
1.2 实验方法
1.2.1 HBA合成途径与宿主适配筛选
1.2.1.1 HBA合成质粒转化不同宿主细胞
将BL21(DE3)、BL21 Star(DE3)、MG1655(DE3)、BW25113(DE3)、JM109(DE3)和shuffie T7-K12六种菌株做成大肠杆菌化学转化感受态,将表达HBA合成途径基因的质粒pET28-HBA[7]分别转化这6种不同的大肠杆菌宿主细胞中,挑取长出的菌落用YZ-RCcobH-F,YZ-RCcobH-R引物进行验证,正确的转化子接入终质量浓度为50 μg/mL卡那霉素的LB液体培养基中培养12~16 h后保藏。
1.2.1.2 菌株生物量的测定
采用紫外分光光度法测定大肠杆菌生物量。取100 μL菌液,用无菌水稀释合适倍数,混匀后测定其在600 nm处的光密度(OD),测量生物量。待发酵结束后再次稀释合适倍数测定大肠杆菌生物量。
1.2.1.3 发酵条件以及发酵样品处理
从保藏这6种大肠杆菌的-80 ℃冻存管中取适量菌液分别划线于含有卡那霉素的LB固体培养基进行活化,挑取新鲜的单菌落于含有卡那霉素的5 mL LB液体培养基中,置于37 ℃、220 r/min摇床振荡过夜培养。按照初始OD600值为0.05接种于含有终质量浓度为50 μg/mL卡那霉素的50 mL TYGly发酵培养基的三角瓶中,在37 ℃,220 r/min条件下培养,当OD600值增长到0.8时加入1 mmol/L诱导剂IPTG,并将温度调至32 ℃诱导T7启动子驱动的基因表达,待32 h发酵结束后进行发酵样品处理。
使用5 mL移液器取发酵液10 mL于15 mL离心管中,4 000 r/min离心10 min弃上清液,并用移液器吸干残留液体。加去离子水,定容至2 mL,混匀菌体。盖好离心管盖后,将离心管放置于灭菌锅100 ℃处理30 min。取出离心管,4 000 r/min离心10 min,转移上清液于1.5 mL EP管中。13 000 r/min离心10 min,用1 mL注射器吸取上清液,通过0.22 μm的水系滤膜过滤至新的1.5 mL EP管。最后,吸取200 μL样品通过HPLC检测HBA的含量。
在发酵3、6、12、18、24、30、36 h取样稀释,测量OD600值,绘制生长曲线。
在发酵6、12、18、24、30、36 h取样700 μL,离心取上清液,通过0.22 μm的水系滤膜过滤至新的1.5 mL EP管。最后,吸取200 μL样品通过HPLC检测乙酸的含量。
1.2.1.4 HPLC定量检测HBA
在安装有Agilent SB-aq柱(4.6 mm×150 mm, 5 μm)的Agilent 1260液相色谱仪上进行分析。样品进样量10 μL。液相条件参考FANG等[7]文献报道。所用流动相成分为:A相为1‰甲酸,B相为含有1‰甲酸的甲醇。流速1.0 mL/min,色谱柱温度35 ℃,检测波长329 nm。梯度洗脱条件:0~2 min,0~25% buffer B;2~4 min,25%~34% buffer B;4~12 min,34%~70% buffer B;12~17 min,70%~100% buffer B;17~23 min,100% buffer B;23~25 min,0~100% buffer B;25~27 min 0% buffer B。
1.2.1.5 HPLC定量检测乙酸
在安装有Bio-Red Aminex HPX-87H柱(7.8 mm×300 mm, 9 μm)的Agilent 1260液相色谱仪上进行分析。样品进样量20 μL。所用流动相成分:C相5 mmol/L H2SO4,流速0.5 mL/min,色谱柱温度60 ℃。
1.2.1.6 酶标仪定量检测ATP
在发酵6、12、18、24、30、36 h取样200 μL,4 ℃、12 000 r/min离心2 min后弃上清液,采用上海碧云天生物技术有限公司ATP检测试剂盒检测,加入400 μL ATP裂解液进行重悬,静置5 min,4 ℃、12 000 r/min离心2 min,吸取上清液50 μL,滴入已在室温放置5 min的含有100 μL ATP检测工作液的Costar 96孔白色不透明板中,立刻混匀用Biotek Synergy HTX酶标仪进行发光检测,根据标准曲线计算样品中胞内ATP的浓度。按照文献方法将OD600转化为细胞干重(dry cell weight,DCW)并将ATP浓度换算成单位细胞干重的摩尔数[16]。
1.2.2 将HBA操纵子整合到基因组
将含有HBA合成途径基因的质粒pCas9-hsdR-HBAD[7]转化目的菌株BW25113(DE3),加入终浓度为10 mmol/L的阿拉伯糖诱导Cas9和λ噬菌体Red重组酶表达,诱导18 h后划线到含有氨苄青霉素和阿拉伯糖的LB固体培养基的板子上,挑取长出的菌落进行验证,验证且测序正确后接入无抗的LB培养基中,转移到42 ℃、220 r/min摇床进行传代培养使质粒丢失,影印后对HBA操纵子整合的位点进行DNA测序,确保整合到目的菌株相应的位点。
1.2.3ackA-pta基因的敲除
根据文献报道的同源重组结合CRISPR-Cas9的方法进行无痕敲除[17]。实验室构建了敲除ackA-pta位点的Cas9质粒[7]。基因组上ackA-pta基因的上下游与相应的Cas9质粒上序列设计成一致。再将相应的Cas9质粒导入到目的菌株化学转化的感受态中,分别挑取单菌落于5 mL含有终质量浓度为100 μg/mL的氨苄青霉素中,在30 ℃、200 r/min摇床培养2 h后,添加终浓度为10 mmol/L的阿拉伯糖进行诱导表达,诱导18 h后划线于含有氨苄青霉素和阿拉伯糖的LB固体培养基的板子上,挑取长出的菌落用YZ-ackA-pta-F/R引物进行验证,验证且测序正确后接入无抗的LB培养基中,在42 ℃、220 r/min摇床进行传代培养使Cas9质粒丢失后保藏。
1.2.4S.meliloti和Z.mobilis两种来源的ED途径的整合
ED途径的整合采用标准化基因编辑方法进行(未发表)。在endA位点引入S.meliloti和Z.mobilis两种来源的ED途径,构建了pDonor01-endA-SmED、pDonor01-endA-ZmED和pCas9g-endA三个质粒。以引入S.meliloti来源的ED途径为例,以MG1655基因组为模板,扩增endA基因的上下游,并胶回收纯化,得到片段endA-up和endA-dn。将endA-up与质粒pMXLK-pFa进行Golden Gate组装,将endA-dn与质粒pMXLK-bFq进行Golden Gate组装,化学法转化大肠杆菌DH5a菌株,蓝白斑筛选涂板后,挑取白色单菌落进行验证,正确的转化子接入终质量浓度为50 μg/mL的卡那霉素中培养12~16 h提质粒并送测序,得到质粒pMXLK-pFa-endA-up和pMXLK-bFq-endA-dn。
以YX18基因组为模板,分别扩增edd、eda基因,胶回收纯化,2片段与质粒pMXLK-aFb进行Golden Gate组装后化学法转化大肠杆菌DH5a,蓝白斑筛选涂板,处理方法同上,得到质粒pMXLK-aFb-SmED。其与pMXLK-pFa-endA-up、pMXLK-bFq-endA-dn和pDonor01共4个质粒进行Golden Gate组装,处理方法同上,得到质粒pDonor01-endA-SmED。引物endA-N20-F、endA-N20-R退火后与质粒pCas9gB进行Golden Gate组装。处理方法同上,得到质粒pCas9g-endA。
向目的菌株中用电转化的方法转入pDonor01-endA-SmED、pCas9 g-endA,挑取单菌落于5 mL含有终质量浓度为100 μg/mL的氯霉素和50 μg/mL卡那霉素中,在30 ℃、200 r/min摇床培养2 h后,添加终浓度为10 mmol/L的阿拉伯糖进行诱导表达,诱导18 h后划线到含有氯霉素、卡那霉素和阿拉伯糖的LB固体培养基的板子上,挑取长出的菌落进行验证,验证且DNA测序正确后丢双质粒后保藏。
2 结果与分析
2.1 氢咕啉酸合成途径与宿主适配筛选
选择合适的宿主是实现目的基因功能表达的关键,大肠杆菌因具有生长快和遗传操作工具成熟等优势而被广泛应用。有文献报道,大肠杆菌是合成维生素B12的前体5-氨基乙酰丙酸(5-aminolevulinic acid,ALA)的优良菌种[18],具有代替传统菌种生产维生素B12的潜力。由于HBA操纵子是T7启动子驱动表达,这里以6种含有T7RNA聚合酶的大肠杆菌作为筛选对象,即BL21(DE3)、BL21 Star(DE3)、MG1655(DE3)、BW25113(DE3)、JM109(DE3)和shuffie T7-K12。将含有HBA合成途径的质粒pET28-HBA[7]通过化学法转化到这6种大肠杆菌中,进行发酵和HPLC液相检测,如图2-a所示。
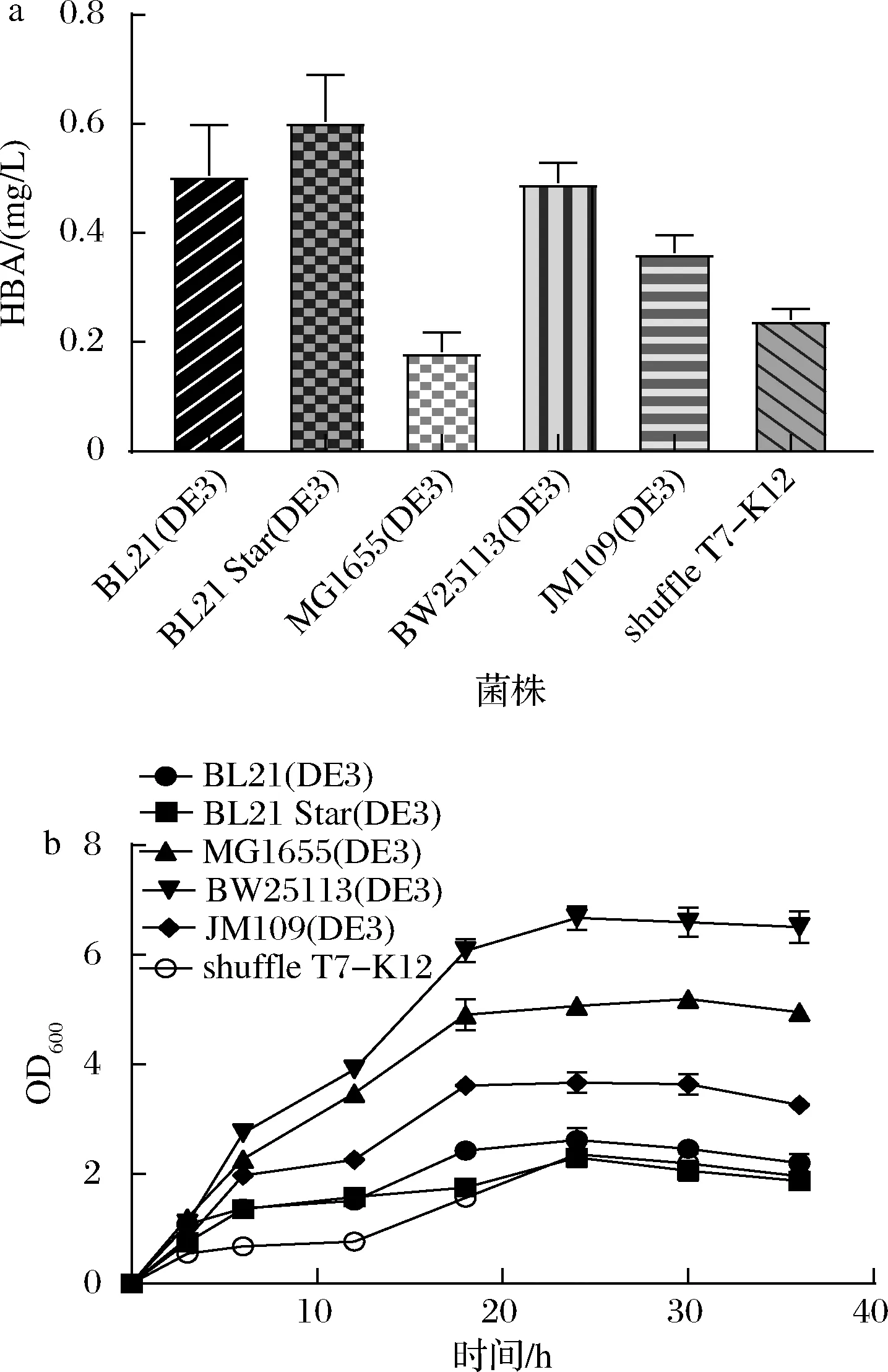
a-不同宿主HBA产量;b-不同宿主生长情况
BL21(DE3)和BL21 Star(DE3)作为宿主合成HBA的产量比较高,但是如图2-b所示,生长曲线显示它们的生物量过低,生物量太低会影响以后工业化生产。MG1655(DE3)作为宿主合成HBA的产量最低。BW25113(DE3)作为宿主的生物量和HBA产量都比较好,HBA的产量可以达到0.49 mg/L,OD600值为6.60。
2.2 HBA操纵子整合到染色体
为了避免质粒表达给菌株造成代谢负担,利用CRISPR/Cas9介导的基因编辑技术将HBA操纵子整合到BW25113(DE3)染色体hsdR位点上,菌株命名为BW01。将BW25113(DE3)和BW01进行发酵以及HPLC检测,如图3所示。BW01的HBA产量得到显著提高,从0.49 mg/L增长到6.38 mg/L,提升了12倍;去除质粒后,细胞生长状态有了明显改善,OD600值从6.60增长到18.58。
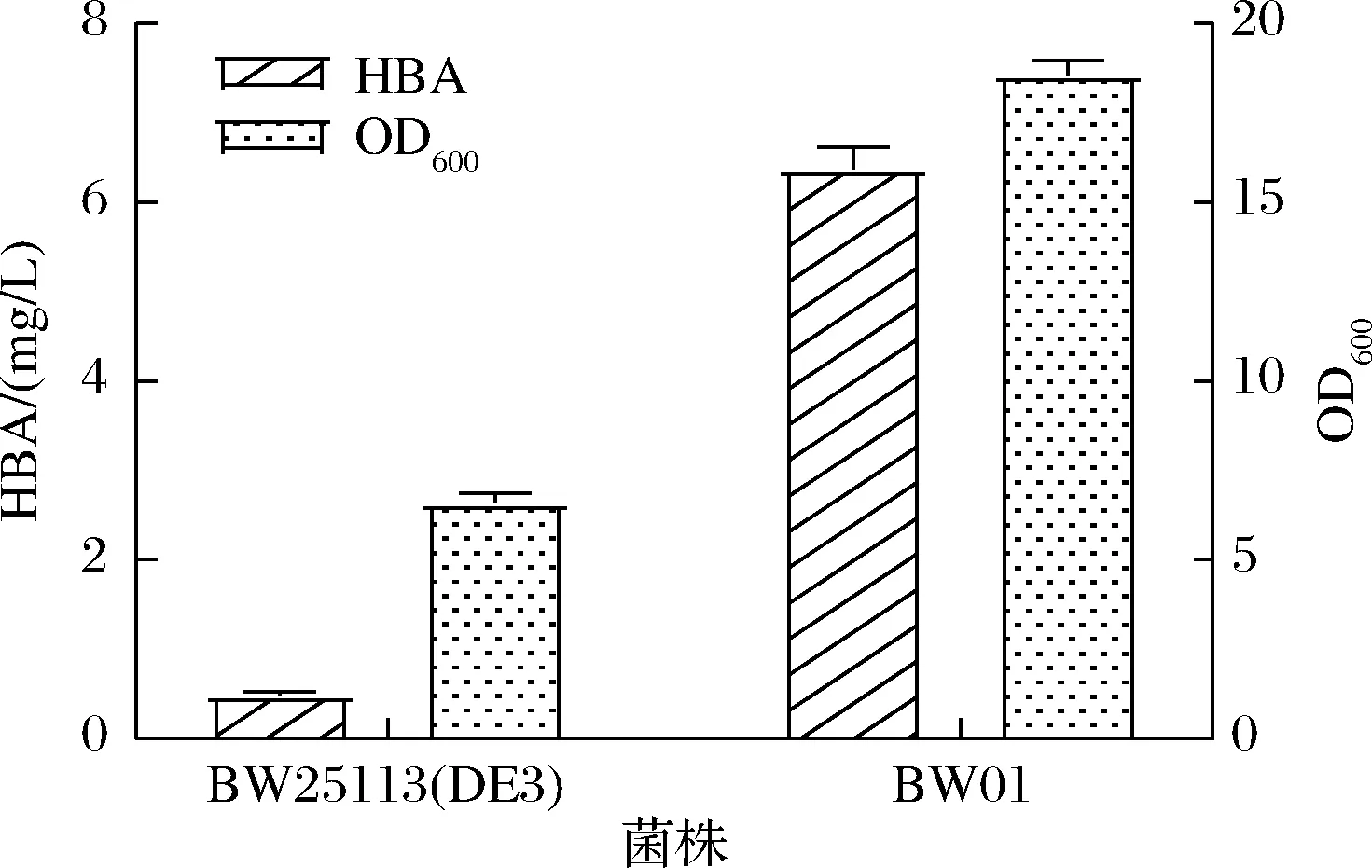
图3 HBA操纵子质粒表达与染色体表达菌株发酵结果Fig.3 Fermentation results of strains HBA operon expressed on plasmid or chromosome
2.3 加强前体供应
在此之前,我们对大肠杆菌进行了生产维生素B12的代谢工程研究,尿卟啉原III是HBA合成的重要前体物质[7]。为避免因工程菌株前体供应不充足,导致HBA的发酵质量浓度以及生产强度低下,通过优化前体尿卟啉原III供应来解除限制,进而提高大肠杆菌HBA合成能力。将生成尿卟啉原III的前体模块整合到基因组PBAD位点,得到菌株BW02,进行发酵以及HPLC检测,如图4所示。HBA的产量从6.38 mg/L增长到11.61 mg/L,提高了82%,OD600值达到19.95。
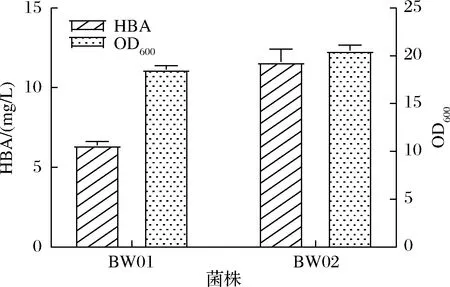
图4 加强前体供应的菌株HBA生产Fig.4 HBA production of strains with enhanced precursor supply
2.4 ackA-pta基因缺失
Pta-AckA途径在大肠杆菌体内初级代谢过程中具有重要的作用[19],生成辅酶A,促进糖酵解和三羧酸循环,是生物体内能量代谢的重要组成部分。中间产物乙酰磷酸可以使丙酮酸转化为乙酸,大肠杆菌好氧发酵时会积累乙酸。乙酸作为副产物持续累积可能对HBA产量有所影响[12]。所以敲除磷酸转乙酰酶基因(phosphotransacetylase,pta)与乙酸激酶基因(acetokinase,ackA),破坏Pta-AckA途径,得到BW03菌株。通过菌株摇瓶发酵和HPLC检测,分析ackA-pta基因的失活对生长代谢和HBA产量的影响,如图5-a和图5-b所示。发现ackA-pta缺失后OD600值略有降低,乙酸生成量大幅降低,BW02、BW03菌株最大乙酸生成量分别为6.82和4.38 g/L。虽然敲除ackA-pta会降低乙酸积累量,但是对HBA合成不利,产量从11.61 mg/L下降到8.65 mg/L,可能因为敲除ackA-pta的同时,腺嘌呤核苷三磷酸(adenosine triphosphate,ATP)供给减少,影响了合成HBA所需前体S-腺苷甲硫氨酸的合成。对2株菌的胞内ATP进行定量,如图5-c所示,发现除了发酵6 h以外,敲除ackA-pta基因后ATP含量的确会有所降低。综合来看,ATP比乙酸对HBA合成影响更大。因此放弃敲除ackA-pta,选择BW02菌株进行后续改造。
2.5 两种来源ED途径的引入
糖酵解(embden-meyerhof-parnas,EMP)途径、磷酸戊糖途径、ED途径和磷酸转酮酶途径是主要的碳代谢途径[20]。ED途径主要由两个反应组成。第一个是由6-磷酸葡萄糖酸脱水酶(6-phosphogluconate dehydrase,EDD)催化,产生2-酮-3-脱氧葡萄糖酸-6-磷酸(2-keto-3-deoxygluconic acid 6-phosphate,KDPG)。第二反应由酮-3-脱氧葡萄糖酸-6-磷酸缩醛酶(keto-3-deoxygluconic acid -6- phosphate acetalase,EDA)催化,产生丙酮酸和甘油醛3-磷酸。与EMP途径相比,ED途径的蛋白质需求减少72%,同时提供的ATP减少一个[21]。EMP途径只能合成NADH,不能合成NADPH,而ED途径可以合成1个NADPH。从ALA每合成1分子维生素B12需要消耗10分子的NADPH[22]。大肠杆菌内源的ED途径过弱,通过引入外源的ED途径有可能提高维生素B12合成中间体HBA产量。
S.meliloti是天然生产维生素B12的菌株,为研究天然生产维生素B12菌株中ED途径的引入对HBA产量的影响,构建了S.meliloti来源的ED途径的质粒。有研究发现,在大肠杆菌MG1655中引入Z.mobilis来源的ED途径,NADPH再生率提高了25倍[23],于是又构建了Z.mobilis来源的ED途径的质粒。因此,选择在BW02菌株endA位点分别整合S.meliloti来源和Z.mobilis来源的ED途径,分别得到菌株BW04和BW05。以BW02菌株为对照菌株,发酵以及HPLC检测,如图6所示。
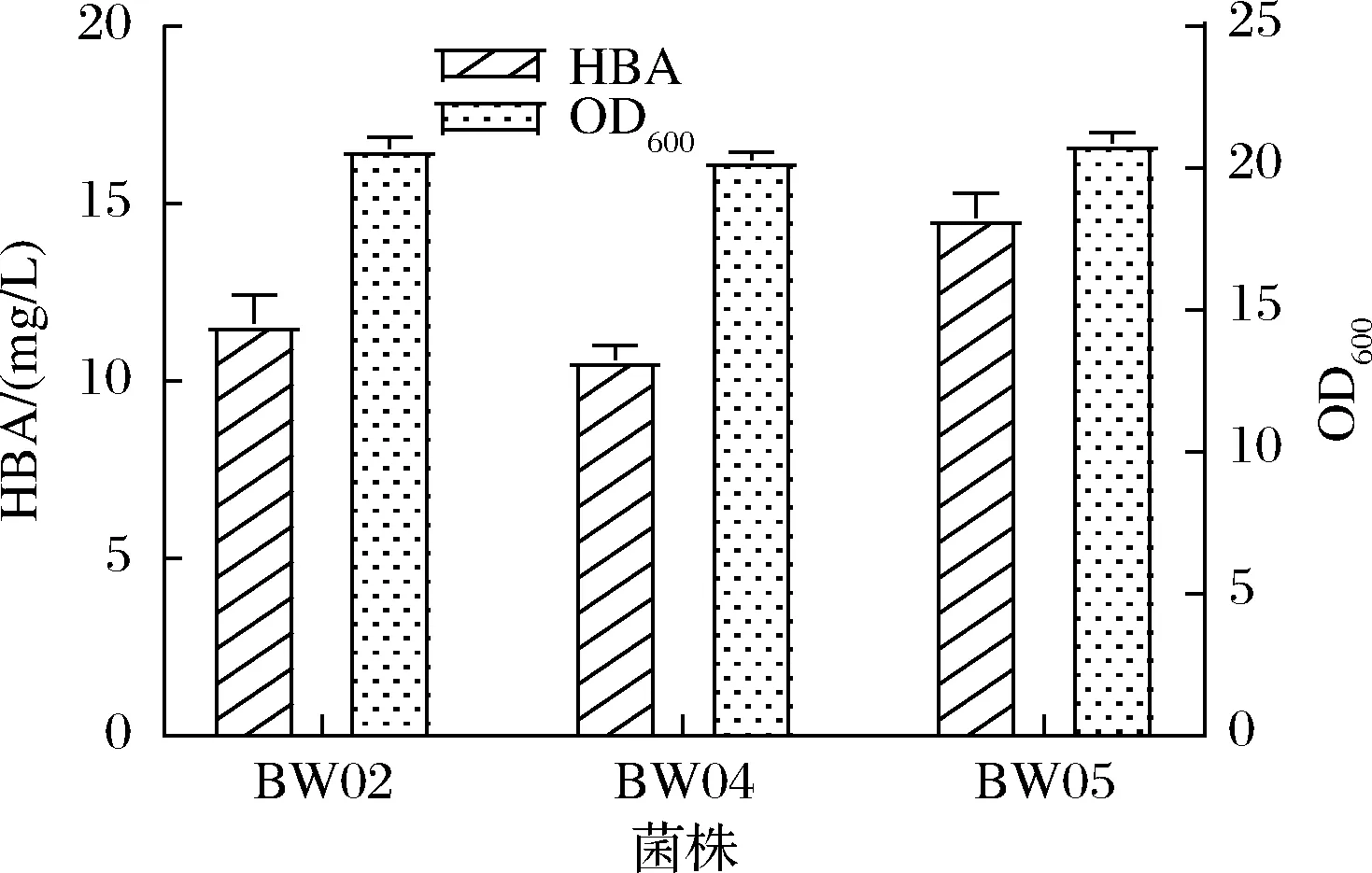
图6 两种来源ED途径引入菌株的HBA生产Fig.6 HBA production of strains with two sources of ED pathways
由图6可知,BW04产量有所下降,从11.61 mg/L下降到10.60 mg/L,分析原因可能是S.meliloti来源的ED途径基因的基因表达水平不平衡导致的[24]。BW05的HBA产量从11.61 mg/L增加到14.60 mg/L。说明Z.mobilis来源的ED途径引入对HBA产量的提高有较大帮助。
3 结论与讨论
大肠杆菌是替代现有维生素B12生产菌种的非常有竞争力的菌种,利用无抗菌株生产维生素B12是未来的发展方向。本文在筛选最佳的合成HBA的宿主BW25113(DE3)基础上采取多个代谢工程策略,整合HBA和前体合成途径基因后HBA产量提高到11.61 mg/L,由于摆脱了质粒对宿主带来的代谢负担,OD600值从6.60提高到20.55。敲除乙酸合成基因ackA-pta不利于HBA合成,表达Z.mobilis来源的ED途径基因后HBA产量最终达到14.60 mg/L。本研究所构建的无质粒重组菌发酵过程中无需添加抗生素,节省了产品分离提取的成本,对生产更有利,具有工业应用潜力。
大肠杆菌好氧发酵有2条产生乙酸途径:一是在乙酸激酶(acetokinase,ACK)和磷酸转乙酰基酶(phosphotransacetylase,PTA)作用下由乙酰CoA生成乙酸,主要在对数期起作用;另一条是在丙酮酸氧化酶的作用下由丙酮酸直接生成乙酸(PoxB途径),主要在对数后期和平台期其作用[25]。直接敲除ackA-pta不利于HBA合成暗示对于乙酸代谢途径的控制要和HBA合成对ATP的需求相协调,这样才能达到既控制了乙酸积累量又提高HBA产量。由于合成HBA需要消耗大量NADPH,引入了Z.mobilis和S.meliloti来源ED途径,但表达S.meliloti来源ED途径基因后HBA产量有所下降,从11.61 mg/L下降到10.60 mg/L,分析原因可能是S.meliloti来源的ED途径基因的基因表达水平不平衡导致的。
本研究在乙酸代谢和ED途径进行了初步改造,接下来工作包括:为避免敲除基因影响过大,精准弱化ackA-pta或poxB基因,平衡乙酸生成和ATP供给;弱化糖酵解途径以提高ED途径代谢流,为维生素B12合成提供充足的NADPH;继续在染色体上整合维生素B12下游合成途径基因获得生产维生素B12的工程菌。
猜你喜欢
中学生数理化·高一版(2022年4期)2022-05-09
天津医科大学学报(2021年1期)2021-12-05
科学(2020年3期)2020-11-26
当代水产(2020年3期)2020-06-15
现代检验医学杂志(2016年5期)2016-08-20
浙江大学学报(工学版)(2016年9期)2016-06-05
当代化工研究(2016年5期)2016-03-20
实用皮肤病学杂志(2015年4期)2015-12-22
医学研究杂志(2015年12期)2015-06-10
茶叶通讯(2014年2期)2014-02-27
