
乳液电纺制备微囊型聚己内酯纳米纤维及其封装蛋白性能
2023-10-17 23:51:08任瑞鹏吕永康
高等学校化学学报 2023年10期
高 冲, 周 全, 杨 帆, 任瑞鹏, 吕永康
(太原理工大学化学工程与技术学院, 省部共建煤基能源清洁高效利用国家重点实验室, 太原 030024)
静电纺丝技术能够在静电场作用下将高分子溶液拉丝制成纳米纤维膜[1], 因其可以环保、 高效、安全地制备纳米材料而备受关注[2,3]. 静电纺丝纳米纤维由于具有大的比表面积和可调节的纤维直径及孔隙率等特性[4,5]而被广泛研究并应用于药物输送[6]、 组织工程[7]、 生物敷料[8]、 固定化酶[9]、 生物传感器[10]和固态电解质[11]等领域.
电纺纳米纤维是封装蛋白质和酶等生物活性分子的绝佳载体, 由于可选择生物相容性高分子和纺丝成膜后方便分离回收的优点, 在固定化酶领域具有良好的应用前景[12,13]. 目前的研究一般是在纳米纤维外表面固定一层生物活性分子, 制备成核壳结构的固定化载体[14~17], 而较少将其固定于纳米纤维内部的微纳结构中[18]. 区室化和微囊泡是在活细胞中发现的细胞器的基本特征, 如线粒体、 叶绿体和高尔基体等, 作为参与级联生化反应的多酶载体, 其确保了生理活动不受外部不良环境的影响[19]. 多年来, 研究者一直致力于用人工合成的类似物来模拟细胞中细胞器的区室化和微囊泡结构[20~23]. 通过脂质体、 水凝胶/纳米纤维包埋或高分子微胶囊来封装生物酶[24~26], 模拟细胞器的微环境, 从而提高酶促反应的效率和选择性[27~29].
通过乳液进行静电纺丝能够将亲水性生物活性分子封装在高分子纳米纤维内部, 并保持这些分子的结构性能和生物活性[3,30,31]. 乳液的制备方法对于材料中形成微囊泡结构至关重要. 乳液由两个或多个不相溶的溶液相组成, 分为连续相和分散相[32]. 根据定义, 油溶性疏水相通常被描述为油相, 另一相为水相; 如果水相分别为连续相或分散相, 则被描述为水包油(O/W)或油包水(W/O)乳液. 为了防止O/W 或W/O 乳液中发生相分离, 一般需要使用乳化剂, 通常有表面活性剂[如十二烷基硫酸钠(SDS)、 聚山梨酯(Tween)和脂肪酸山梨坦(Span)]、 无机纳米颗粒(如碳酸钙、 二氧化硅)和生物大分子(如多糖类的明胶、 海藻酸, 蛋白质类的大豆蛋白、 乳清蛋白)等[33]. 添加适当且足量的乳化剂能够有效稳定乳液, 使两相混合形成有利于纳米纤维包封的微纳结构[34]. 此外, 随着可再生新材料的开发,生物相容性高分子聚合物[如聚乙烯醇(PVA)、 聚环氧乙烷(PEO)和聚己内酯(PCL)等]也被用于进行乳液静电纺丝[35]. 但通过乳液形成的微囊泡结构在静电纺丝过程中受电场力的作用会发生破乳现象,囊泡之间相互融合并沿纤维轴向拉伸, 最终形成中空/核壳结构的纳米纤维, 达不到模拟细胞器微纳结构的效果.
本文选择在PCL 溶液中添加两亲性三嵌段共聚物泊洛沙姆Pluronic®F108(PEO-PPO-PEO)作为乳化剂, 探索了通过超声和水相添加剂等方法形成长时间稳定的载酶W/O乳液的条件, 同时通过控制纺丝条件, 解决了常规乳液纺丝过程中产生的破乳问题, 最终制备出微囊型PCL纳米纤维膜作为固定化酶的载体(Scheme 1), 通过荧光标记的牛血清白蛋白(BSA)验证了蛋白被有效封装在纳米纤维内部的微囊泡结构中, 并研究了其在缓冲液中的释放行为, 为构建高效仿生的载酶纳米材料提供了思路.
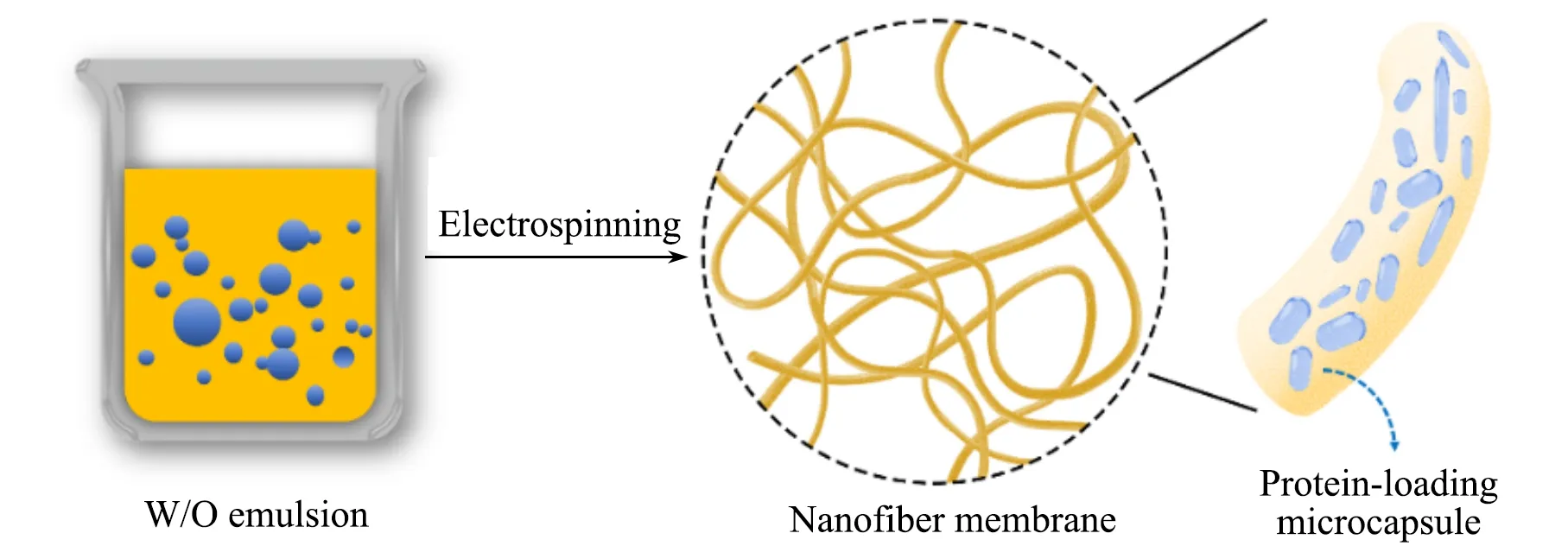
Scheme 1 Microcapsular structure of nanofibers for protein encapsulation by emulsion electrospinning
1 实验部分
1.1 试剂与仪器
聚己内酯(PCL,Mw=100000), 广东华创塑化有限公司; 泊洛沙姆Pluronic®F108(Mn=14600)和二甲基亚砜(DMSO, 分析纯), 上海阿拉丁生化技术有限公司; 二氯甲烷(DCM, 分析纯)、 多巴胺盐酸盐(DA)、 甘油(Gly, 纯度≥99%)、 牛血清蛋白(BSA)、 Bradford 试剂盒和异硫氰酸罗丹明B 荧光素(RBITC), 上海麦克林生化科技有限公司.
JY92-IIN 型超声波细胞破碎仪, 宁波新芝生物科技股份有限公司, 设备最大功率为650 W; MX-S型台式混匀仪, 北京大龙兴创实验仪器股份公司; 752型紫外-可见分光光度计(UV-Vis), 上海频谱仪器有限公司; JY-82型接触角测量仪, 承德鼎盛试验机检测设备有限公司; AutoPoreV 9620型高性能全自动压汞仪, 美国Micromeritics 公司; TCS SP8 型激光共聚焦扫描显微镜(CLSM), 德国Leica 公司;Gemini 300型扫描电子显微镜(SEM), 德国Zeiss公司; Nova 4000e型全自动比表面积及孔隙度分析仪,美国Quantachrome公司; 5944型电子万能材料试验机, 美国Instron公司.
1.2 实验过程
1.2.1 稳定W/O 乳液的制备 将一定量PCL 溶于二氯甲烷中并搅拌4 h, 配制成10%(质量分数)的纺丝液, 分别加入10%, 20%和30%(相对于聚合物的质量分数)的F108, 待其溶解后再分别加入10%,20%和30%(相对于二氯甲烷的体积分数)的水, 在冰浴中进行超声处理, 设置超声波细胞破碎机功率分别为260, 325, 390, 455 W, 每超声5 s 后, 空5 s(占空比1∶1), 超声总时间分别为30, 60, 90 和120 s, 形成稳定的W/O乳液并静置观察. 另外在水相中添加20%(体积分数)甘油(Gly)、 2 mg/mL多巴胺盐酸盐(DA)和20%(体积分数)Gly+2 mg/mL DA分别测试乳液的稳定性.
1.2.2 静电纺丝液的配制和纳米纤维膜的制备 静电纺丝液为10%(质量分数)PCL中添加30%(质量分数)的F108和10%(体积分数)的水相, 在超声功率为390 W时超声90 s形成的乳液. 水相分别为添加与未添加20%(体积分数)Gly+2 mg/mL DA的高纯水. 分别将纺丝液装入20 mL注射器中, 针头内径为0.51 mm, 辊轴接收器与针头距离为15 cm, 转速为100~500 r/min, 设置流速为0.8~1.5 mL/h, 调节电压至 10~15 kV, 在室温(30 ℃), 湿度40%环境下纺丝5 h后, 收集纳米纤维膜. 另外, 在相同实验条件下用平板接收器收集纤维膜.
1.2.3 BSA 的荧光标记和蛋白的定位分布 将50 mg BSA 溶于10 mL 0.1 mol/L pH=7.4 的磷酸盐缓冲液中. 将10 mg异硫氰酸罗丹明B溶于1 mL DMSO中, 取400 μL该溶液用0.5 mol/L pH=9的碳酸盐缓冲溶液稀释至1 mL, 然后逐滴滴加到BSA溶液中, 冰浴条件下搅拌4 h. 反应完成后, 以4000 r/min的速度离心10 min, 用PD-10/G-25柱子脱盐, 于4 ℃保存. 将荧光标记和未标记的BSA 进行聚丙酰胺凝胶电泳(SDS-PAGE), 然后在紫外灯下照射观察BSA的荧光标记情况.
将乳液中的BSA用相同数量的荧光标记的BSA代替, 即在水相中添加荧光标记的20 mg/mL BSA+20%(体积分数)Gly+2 mg/mL DA 作为纺丝液, 在1.2.2节中的静电纺丝条件下进行纺丝, 收集的膜用CLSM观察荧光分布.
1.2.4 载BSA 纳米纤维中蛋白的释放实验 水相为添加与未添加20%(体积分数)Gly+2 mg/mL DA 的含有20 mg/mL BSA的溶液, 按照1.2.2节中的纺丝条件制备纳米纤维膜. 将载BSA的纳米纤维膜浸泡在pH=7.4 的Tris-HCl 缓冲液中, 放置在30 ℃、 转速100 r/min 的摇床内. 分别在0, 10, 20, 30 min 和1, 2, 4 h各取3 mL蛋白释放液, 采用考马斯亮蓝法测蛋白浓度, 通过紫外-可见分光光度计在595 nm波长处测定溶液的吸光度(A595), 根据BSA蛋白标线(图S1, 见本文支持信息)计算溶液中蛋白浓度, 绘制出蛋白释放曲线.
溶液中BSA蛋白浓度(c, μg/mL)按BSA标线进行计算:
式中:C(μg/mL)为缓冲液中蛋白浓度; 相关系数R2=0.998.
蛋白释放率(Release rate, %)按下式计算:
式中:V(mL)为缓冲液体积;m(μg)为样品膜中理论蛋白质量(膜中理论蛋白质量=纺丝液中添加的蛋白总质量/纺丝后膜的总质量×样品膜的质量).
1.3 测试与表征
1.3.1 乳液的形貌结构表征 将乳液在液氮中冷冻5~10 min, 然后冷冻干燥4 h, 形成固体. 将该固体剪碎, 用导电胶将碎末粘贴在样品台上, 待纤维膜表面喷金处理后, 利用扫描电子显微镜观察其形貌. 利用ImageJ软件测量获得孔径分布图. 为了进一步验证乳液囊泡直径大小, 将处理后的乳液进行氮气吸附-脱附实验, 采用BET法和压汞法获得冷冻干燥后乳液的孔体积和孔径数据.
1.3.2 纳米纤维的形貌结构表征 用导电胶将纳米纤维膜裁剪成小块的待测样品, 粘贴在样品台上,待纤维膜表面喷金处理后, 利用扫描电子显微镜观察其形貌. 利用ImageJ软件测量获得纤维的平均直径和直径分布图. 将纳米纤维膜剪碎, 真空干燥24 h, 使用BET 法和压汞法获得比表面积、 孔体积和孔径等数据.
1.3.3 纳米纤维膜的亲水性测试 将纤维膜剪成2 cm×2 cm大小, 采用接触角测量仪拍摄记录水滴与纤维膜的接触角.
1.3.4 纳米纤维膜的力学性能测试 将纤维膜裁剪成80 mm×20 mm 的样条, 在纤维膜上随意取3 个点, 用厚度仪测定其厚度, 取平均值, 即为纤维膜厚度. 将纳米纤维膜置于抗拉强度测量仪上, 在室温下进行拉伸实验.
2 结果与讨论
2.1 稳定W/O乳液的形成条件
2.1.1 乳化设备选择 不同的乳化设备由于作用方式和设备功率的影响, 对乳液中的液滴大小分布和乳液的稳定性都会有明显的影响. 实验中对比了台式混匀仪和超声波细胞破碎仪对乳液形成的影响.混匀仪采用被动传递振动的方式进行乳化, 需要用手将装有乳液的试管紧紧压在震动盘上, 设备输出功率为10 W, 以3000 r/min的频率做圆周振荡, 混匀5 min后取一部分乳液快速冷冻干燥后观察形貌.由图1(A)可见, 混匀仪形成的囊泡较稀疏且分布不匀, 统计分析囊泡的平均直径为(7.557±2.176) μm[图1(C)], 并且形成的乳液静置30 min后出现明显的分层现象[图S2(A)和(B), 见本文支持信息].

Fig.1 SEM images of emulsion after lyophilization(A, B) and statistical distribution of microcapsule diameter(C, D)
采用超声波细胞破碎仪进行乳化, 将超声探头直接伸入乳液中进行超声振荡, 基于超声波在液体中的空化效应形成的高频交变水压强和剪切力达到乳化的效果. 设备总功率650 W, 以5 s为周期、 占空比1∶1进行间歇工作, 由于换能器直接传递能量效率高, 必须在冰水浴中进行超声处理以保证乳液不会剧烈升温. 超声后形成的W/O乳液在静置24 h后未观察到分层现象[图S2(C)~(D), 见本文支持信息]. 将其冷冻干燥后进行SEM观察, 发现形成了大量均匀分布的囊泡, 区室化结构显著[图1(B)],统计分析表明囊泡的平均直径为(7.025±2.347) μm[图1(D)]. 综合来看, 采用超声方式形成的乳液均质化程度更高, 形成的囊泡分布更均匀, 稳定时间更长.
2.1.2 超声参数 通过调整超声细胞破碎仪的功率和超声时间, 探究其对乳液形貌和结构的影响. 改变超声功率比分别为260~455 W, 观察不同超声功率下乳液冷冻干燥后的形貌(图S3, 见本文支持信息). 结果表明, 随着超声功率增大, 乳液中囊泡的直径逐渐变大, 由颗粒状、 粘连少孔状逐渐形成区室化多孔状态. 当超声功率达到390 W时, 乳液中囊泡密集且分布均匀, 呈现明显的区室化和多孔结构; 而当超声功率超过390 W后, 乳液中囊泡的直径明显变大, 形态由偏圆形被拉长形成贯通孔状, 说明超声功率过大时, 乳液被打破、 囊泡之间发生了融合.
控制超声总时间分别为30~120 s, 超声5 s后间隔5 s, 观察不同超声时间下乳液的形貌(图S4, 见本文支持信息). 发现随着超声时间的增加, 乳液中颗粒形态减少, 囊泡分布均匀, 与前面调整超声功率由260 W增大到455 W导致的乳液形貌变化现象一致. 当超声时间超过120 s后, 乳液中的囊泡壁上出现了很多小孔, 说明超声时间过长, 出现了双乳液现象, 最终形成了小孔和大孔交错分布的形貌, 造成纺丝过程中纤维强度不高, 容易断裂. 上述结果表明, 在390 W超声功率下超声90 s能够形成稳定的区室化和多孔结构的乳液.
2.1.3 水添加量 水的添加量会直接影响乳液中可溶解生物活性分子的量, 因此应提高乳液中水的占比; 但水含量过高时很难进行乳液静电纺丝, 这是由于乳液中溶解高分子的油相和溶解蛋白/酶分子的水相之间的黏度差别过大时会在纺丝过程中发生相分离, 导致破乳, 无法形成连续的纳米纤维. 逐步提高乳液中水的添加量[分别从5%, 8%, 10%, 20%提高到30%(体积分数)], 采用SEM观察乳液冷冻干燥后的形貌(图2). 当水添加量达到10%时, 可以看到乳液内部有许多囊泡, 且孔径分布一致; 当水添加量为20%时, 乳液内部不仅形成囊泡, 在囊泡周围还存在许多不同大小的球形颗粒; 当水添加量为30%时, 乳液内部观察不到明显的囊泡结构, 在贯通的孔周围附着了大量的球形颗粒. 表明当纺丝液和水相体积比超过9∶1后, 在超声过程中乳液的均质化程度大大降低, 发生破乳并进一步导致在水相中形成了油相囊泡的双乳液现象, 从而在乳液冷冻干燥后观察到球状的高分子颗粒. 故水添加量为10%(体积分数)时为最佳, 可较好地满足乳液纺丝的要求.

Fig.2 SEM images of the emulsions prepared with different addition amounts of water
2.1.4 乳化剂添加量 乳化剂的选择和添加量对乳液的形成和稳定性起重要作用. 实验选用两亲性非离子表面活性剂Pluronic®F108作为乳化剂, Pluronic®F108是由PEO和PPO组成的ABA型三嵌段共聚物, 平均分子量Mn=14600, 其中亲水段PEO占总分子量的82.5%, 具有较大的亲水亲油平衡值(HLB>24), 对蛋白类生物分子有较好的相容性, 可以有效降低混合体系的界面张力, 有利于形成稳定的乳液. 逐步提高Pluronic®F108的添加量(质量分数), 考察其对乳液形貌和稳定性的影响. 从图3可以看出, 当乳化剂添加量为10%时, 乳液内部呈粘连状, 孔分布不均匀; 当乳化剂添加量为20%时, 粘连态减少, 变成明显的球状颗粒相互连接, 表明此时乳化剂添加量较少, 不足以使乳液稳定; 当乳化剂添加量为30%时, 可以看到紧密相连的囊泡, 形成区室状多孔结构, 孔分布均匀, 孔径一致性高; 而当乳化剂添加量超过30%时, 乳液更容易产生泡沫, 不利于纺丝. 提高乳化剂比例, 油相中可以形成致密的聚合物网络, 抑制了内水相之间的聚集和内水相向外水相的扩散, 乳液的稳定性显著提高[36], 当乳化剂浓度为30%(质量分数)时, 可以形成长时间稳定的乳液.

Fig.3 SEM images of the emulsions prepared with different addition amounts of Pluronic® F108 emulsifier
2.1.5 水相中添加剂对乳液和纺丝的影响 通常制备乳液时, 在水相中不需要额外的添加剂; 但为了减少乳液在纺丝过程中因两相黏度差异导致的相分离问题, 需要选择适当的水相添加剂, 增加乳液的黏度, 从而在微囊泡中原位固定化蛋白类生物分子且具有一定的保护作用. 多巴胺(DA)可以自发地氧化聚合, 形成具有粘附性的聚多巴胺涂层, 能够牢固地附着在几乎所有类型的材料表面上[37]. 在乳液的水相中添加多巴胺可以在囊泡内原位聚合生成聚多巴胺层, 从而有助于蛋白的固定化. 而水相中添加甘油(Gly)可以对蛋白类生物分子起保护作用, 减少有机溶剂的变性作用, 同时增加乳液中水相的黏稠度, 有利于后续的纺丝过程. 在水相中分别添加20%(体积分数)Gly、 2 mg/mL DA和同时添加Gly与DA(图4), 乳液中囊泡的形貌变化不大, 仍然呈区室状多孔结构, 但同时添加甘油和多巴胺的样品微囊泡的大小分布更均匀, 孔边缘更光滑清晰.

Fig.4 SEM images of the emulsions prepared with different water phase additives
2.2 微囊型纳米纤维的形貌和结构分析
2.2.1 纳米纤维的表面形貌 选择具有生物相容性的高分子PCL作为乳液的油相, 对比了在水相中添加与不添加甘油和多巴胺的乳液得到的静电纺丝纳米纤维的形貌(图5). 由水相中未添加甘油和多巴胺制备的乳液纺丝的SEM照片[图5(A)和(C)]可以看出, 纤维表面附着有一些不规整的小颗粒, 纤维之间明显粘连, 这可能是因为电纺过程中存在一定程度的乳液相分离, 出现了一些聚合物胶束和囊泡融合现象; 同时纤维直径差异较大, 说明存在不规则电纺射流. 统计分析图[图5(A)]中纳米纤维的直径分布[图5(B)]可知, 纤维的平均直径为(1.894±0.658) μm, 直径最小为0.831 μm, 最大为4.43 μm. 而由水相中添加了甘油和多巴胺的乳液得到的纳米纤维表面光滑平整且直径分布比较均匀[图5(D)], 说明纺丝液黏度增大, 乳液射流在电场力的作用下被更充分地拉伸, 并且聚合物分子链沿射流轴的取向更加均匀[38]. 此外, 实验中采用低转速的辊轴接收器收集纳米纤维, 因为过高的接收转速会在电场力存在的情况下额外增加机械拉伸, 进一步加强纳米纤维的轴向取向, 导致纤维内部的微囊泡发生融合和破乳, 从而最终形成核壳型纳米纤维. 因此, 为保持乳液中微囊泡的存在, 应尽量控制较低的辊轴转速(100~500 r/min)或者采用平板接收器.

Fig.5 SEM images(A, C, D) of nanofiber membranes prepared by emulsion electrospinning and statistical distribution of nanofiber diameter from (A)(B)
2.2.2 纳米纤维膜的表面特性和力学性能 在纺丝乳液的水相中添加甘油和多巴胺会对纳米纤维的表面特性和力学性能产生影响. 如图S5(见本文支持信息)所示, 在水相中添加甘油和多巴胺后, 纳米纤维膜的水接触角从64.61°增加到110.53°, 表明水相添加剂改变了纳米纤维膜的表面性质, 其疏水性极大增加. 由于添加甘油和多巴胺后乳液更加稳定, 在静电纺丝过程中不会出现明显的破乳现象, 因此纳米纤维表面更多地呈现出高分子PCL 本身的疏水性; 而未加入水相添加剂的对照组显现出亲水性, 间接证明了其纺丝过程中可能存在相分离问题. 水相添加剂也会提高纳米纤维膜的力学性能(图S6, 见本文支持信息), 使其抗拉强度从4.19 MPa提高到4.80 MPa, 拉伸形变率从48.68%提高到53.12%, 纳米纤维膜具有了更好的韧性.
2.2.3 纳米纤维膜的孔径分布 为了证明乳液和纳米纤维中存在微囊泡, 采用氮气吸附-脱附实验和压汞法两种方法测试材料内部的孔隙. 由图6可见, 乳液经冷冻干燥后测试氮气吸附得到的孔径分布[图6(A)]与纳米纤维膜的孔径分布基本一致[图6(B)], 集中在3.17 nm左右, 纤维膜还存在6.22 nm左右的微孔, 并且计算得出纳米纤维膜的比表面积为31.784 m2/g(图S7, 见本文支持信息). 这些数据与通过SEM图片直接测量的孔径分布数值相比明显偏小, 这是由于直接观察法受视野限制只能反映极小范围的样品情况, 而间接测量法如氮气吸附可以反映的样品尺度大, 更代表整体情况; 但是采用BET法分析氮气吸附-脱附实验结果是依据氮气的毛细管凝聚估算材料的孔径分布, 会低估中孔和大孔的含量, 只能给出比较准确的比表面积和微孔含量, 因此适合表征2~50 nm的孔径. 考虑到乳液和纳米纤维中的微囊泡尺寸较大且呈现闭孔状态, 很可能会影响氮气吸附的实验结果, 因此采用压汞法对样品实施进一步测试. 压汞法属于侵入式间接测量法, 通过测量注汞/退汞的体积和压力变化计算出孔径, 适合表征>50 nm的孔径, 高压下可以破坏侵入微囊泡内部, 实验结果相对更准确. 结果表明, 乳液中微囊泡的孔径主要分布在2.5 μm左右[图6(C)], 还有少量24.2 μm左右的孔, 与SEM观察数据比较一致; 而纳米纤维膜内微囊泡的孔径集中分布在1.06 μm左右[图6(D)]. 此外, 通过压汞法测试可以进一步计算得到纳米纤维膜材料的孔隙率为68.37%.
2.3 载BSA纳米纤维中蛋白的定位与释放行为
2.3.1 荧光标记BSA 在纳米纤维中的分布特性 为了进一步验证纳米纤维中存在微囊泡并可以封装蛋白质/酶等生物活性分子, 在乳液制备时向水相中添加荧光分子标记的牛血清白蛋白(BSA)作为模型蛋白, 通过CLSM观察纳米纤维内部蛋白的定位和分布情况. 通过激光激发标记荧光分子的BSA产生红色荧光, 发现3组照片中明场下的纤维图像和暗场下的点状荧光图像可以有效重合(图7), 直观地表明纳米纤维内部存在点状分布的BSA, 进一步证明了纤维内部存在着封装蛋白的微囊泡结构. 同时,由于静电纺丝过程中的电场拉伸作用和溶剂蒸发导致纳米纤维内部的微囊泡沿纤维轴向取向, 不是呈完美的球形而是呈长棒状[见图7(B), 7(E)和7(H)中白色箭头所标注的区域]. 统计分析表明, 纤维内的微囊泡平均轴向长(1.005±0.399) μm, 径向宽(0.471±0.153) μm, 与2.2.3节中采用压汞法测量纳米纤维膜内部孔径得到的1.06 μm一致. 此外, 在图7中也发现纳米纤维内部存在部分微囊泡破乳并相互融合形成核壳结构的现象[见图7(E)和7(H)中白色圆圈所标注的区域], 这可以通过适当降低纺丝电压, 减少电场力拉伸作用和采用平板接收器消除机械应力等方法得以改善.

Fig.7 CLSM images of nanofiber membrane of different sites with red fluorescent labeled BSA
2.3.2 载BSA 纳米纤维的体外蛋白释放行为 通过测试载BSA 纳米纤维的体外蛋白释放行为和蛋白负载量, 也可以间接证明蛋白被固定在纳米纤维内部的微囊泡中. 在水相中添加20 mg/mL 高浓度的BSA, 同时对比甘油和多巴胺两种水相添加剂的固定化效果. 将制得的纳米纤维在pH=7.4的Tris-HCl缓冲液中浸泡4 h, 通过考马斯亮蓝染色-紫外吸光法测试纳米纤维内部封装蛋白的释放行为. 由图8可见, 水相中不含甘油和多巴胺添加剂的样品, 在10 min 内就快速达到最大释放量, 蛋白释放率为14.83%并且随后一直保持稳定, 说明纤维中存在较少量的微囊泡破乳现象, 导致流失的蛋白快速释放出来, 剩余的85.17%的BSA被固定在纳米纤维的微囊泡内. 水相中额外加入甘油和多巴胺的样品蛋白释放率在120 min 达到10.03%并保持稳定, 表明89.97%的BSA被封装在纳米纤维内部的微囊泡中,蛋白的固定化率进一步提高了4.8%, 通过计算得出纳米纤维内BSA 的负载量可达到12.89 mg/g. 由于水相中多巴胺的加入, 可以在微囊泡中原位聚合形成聚多巴胺层固定化蛋白, 最终减缓了蛋白释放速率并明显降低了BSA 的释放量, 表明微囊型纳米纤维可以有效固定化酶, 减少蛋白流失并提高蛋白的封装率, 从而增加酶的重复使用性.

Fig.8 Protein release rate curves of nanofiber membrane
3 结 论
采用超声乳化方法, 通过调节水添加量、 乳化剂添加量和水相添加剂等参数, 制备了长时间稳定的W/O 乳液, 经乳液静电纺丝制备了封装BSA 的微囊型PCL 纳米纤维膜. 最佳的乳液配制条件为:10%(质量分数)PCL纺丝液中添加30%(质量分数)乳化剂Pluronic®F108, 水相中添加20%(体积分数)甘油、 2 mg/mL多巴胺和20 mg/mL蛋白质/酶, 控制纺丝液和水相体积比为9∶1, 超声功率390 W, 5 s间歇超声90 s(占空比1∶1). 纺丝过程中采用较低电压和平板接收器, 制备的纳米纤维内部呈现区室化微囊泡结构, 比表面积为31.784 m2/g, 内部微囊泡孔径集中分布在1.06 μm, BSA 负载量可达12.89 mg/g, 4 h的蛋白释放率仅为10.03%, 蛋白的固定化率高达89.97%, 可以有效在材料内部微囊泡中封装酶蛋白, 是一种不同于常规中空/核壳结构的仿生功能化微囊型纳米纤维材料, 有望在固定化酶和生物催化等领域发挥出巨大潜力.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/20230199.
猜你喜欢
九江学院学报(自然科学版)(2022年2期)2022-07-02 02:33:28
云南化工(2021年7期)2021-12-21 07:27:36
石油地质与工程(2019年3期)2019-09-10 08:27:54
中国钼业(2019年2期)2019-01-19 15:54:06
水利技术监督(2016年6期)2017-01-15 14:01:33
燕山大学学报(2015年4期)2015-12-25 02:19:46
合成技术及应用(2015年3期)2015-12-11 08:36:27
西北园艺(果树)(2015年1期)2015-02-21 16:44:50
湿法冶金(2014年3期)2014-04-08 01:04:51
癌变·畸变·突变(2014年2期)2014-03-01 04:39:43
