
醛酮树脂基非金属催化剂催化氧气氧化苯制备苯酚
2023-10-14 07:52王伟涛鲍婷玉姜旭禄何珍红王宽杨阳刘昭铁
化工进展 2023年9期
王伟涛,鲍婷玉,姜旭禄,何珍红,王宽,杨阳,刘昭铁,2
(1 陕西科技大学化学与化工学院,陕西 西安 710021;2 陕西师范大学化学与化工学院, 陕西 西安 710119)
苯酚是重要的化学品和中间体,广泛应用于各种醛类树脂和酚类衍生物等化学品的工业合成,其在化工生产中起着举足轻重的作用[1-2]。工业生产苯酚主要以苯作为原料,采用异丙苯三步法制备苯酚,但该工艺存在污染大、能耗高和产物选择性低等缺点[3-5]。因此,氧气直接氧化苯制备苯酚作为绿色的苯酚制备路线,近年来得到了广泛的研究。氧气直接氧化苯制备苯酚反应一般以Cu、Fe 和V等具有氧化还原活性的金属作为催化剂[6-8]。然而,此类催化剂成本较高且存在活性金属易发生浸出和团聚的缺点[9-10]。相比之下,使用以碳材料(如碳纳米管、碳纤维和石墨烯等)为主的非金属催化剂可以有效避免这一问题。碳材料以其较高的稳定性、良好的形貌可控性和丰富的表面性质而广泛应用于催化领域[11-14],但其在苯氧化制备苯酚反应中的应用研究较少。王军课题组[15-17]通过煅烧法制备的介孔碳材料、硝酸处理的活性炭、水热法制备的碳材料等表面具有丰富含氧基团的碳材料,催化氧气氧化苯得到10.0%~13.1%的苯酚产率,推测催化剂表面的酮羰基和酚羟基可能是催化活性位点。本文作者课题组[18-19]也发现具有丰富含氧官能团的亲水性碳材料对氧气氧化苯制备苯酚具有催化活性;通过水热法制备的醌胺聚合物可以催化该反应得到15.9%的苯酚产率,同时推测催化剂中的醌/氢醌基团是其催化反应的活性位点。基于此,本文设想能否设计一种具有丰富酮C= = O 基团的材料作为非金属催化剂,实现非金属催化氧气氧化苯制备苯酚。
醛酮树脂是酮与醛经缩聚而成的高分子聚合物,其表面含有丰富的酮C= = O 基团,这有望为苯与氧气直接羟基化制备苯酚反应提供催化活性。因此,本文以醛酮树脂作为对象,利用简单水热法制备得到改性的醛酮树脂基催化剂。通过对合成的催化剂进行系列表征,研究了催化剂表面形貌和表面官能结构;对反应条件进行优化,所得苯酚的最佳产率为16.3%。此外,还提出了醛酮树脂基非金属催化剂催化苯氧化制备苯酚的反应机理。
1 实验部分
1.1 实验试剂
苯,分析纯,上海泰坦科技股份有限公司;环己酮,分析纯,上海阿拉丁试剂有限公司;乙二醛溶液,分析纯,上海麦克林生化有限公司,C2H2O2含量不少于40%;无水乙酸锂、氢氧化钠,分析纯,上海麦克林生化有限公司;环戊酮,分析纯,阿达玛斯试剂(上海)有限公司。
1.2 实验仪器
高压反应釜和TRC-2 恒温调速磁力搅拌器,安徽华蕊实验设备有限公司;依利特3100 型高效液相色谱仪,DAD3100 二极管阵列检测器,大连依利特分析仪器有限公司;电热鼓风干燥机,上海科恒实业发展有限公司;BSA224S型电子天平,赛多利斯科学仪器(北京)有限公司;SHZD(Ⅲ)型循环水式真空泵,予华仪器(巩义)有限公司。
1.3 催化剂制备
通过水热法制备了环己酮-乙二醛树脂基催化剂(HCG),具体步骤如下:在250mL 三颈烧瓶中加入0.08mol环己酮、0.3mol乙二醛和10mL去离子水,搅拌下在油浴中升温至65℃,在搅拌下逐滴加入1.2mL 30%的NaOH 溶液,将溶液升温至85℃后继续搅拌并回流20min。将所得溶液转移至带有50mL聚四氟乙烯内衬的水热釜中,在200℃下水热5h。水热过程完成后滤出固体物并用去离子水和乙醇洗涤,之后在80℃干燥过夜,将所得催化剂命名为HCG。以0.08mol 环戊酮和0.3mol 乙二醛为前体,其他制备步骤和条件均与HCG 相同,将所得催化剂命名为HPG。
1.4 催化剂表征
使用型号为Rigaku Ⅳ的X 射线衍射仪(XRD)对催化剂进行表征,配备Cu靶,陶瓷X光管,扫描范围5°~80°,扫描速率为5°/min。使用日立S-4800型扫描电子显微镜(SEM)对催化剂的形貌进行观测。使用VECTOR-22 型傅里叶红外光谱仪(FTIR)对催化剂的官能团进行测定,采用KBr 压片法制样,波数范围为400~4000cm-1。催化剂的孔结构(BJH)和比表面积(BET)在液氮-196℃环境中使用Gemini Ⅶ 1239型比表面积分析仪进行分析。采用型号为Thermo Scientific K-Alpha+的X 射线光电子能谱仪(XPS)对催化剂表面各元素的状态和含量进行测试。
1.5 催化性能测试
在10mL高压反应釜中加入苯(5mmol)、无水乙酸锂(0.4g)、70%(体积分数)乙酸水溶液(3.0mL)和100mg 催化剂;充入3.5MPa O2,在反应温度下搅拌至反应时间。反应结束后将催化剂离心分离,反应液用甲醇定容到25.0mL。采用依利特3100 型高效液相色谱仪对反应液进行检测。该反应的副产物主要有对苯二酚、对苯醌和二氧化碳。
1.6 自由基猝灭实验
以5mmol 叔丁醇(TBA,·OH 清除剂) 或5mmol 2,6-二叔丁基对甲酚(BHT,·O-2清除剂)进行自由基猝灭试验。在苯(5mmol)、自由基清除剂、HCG(0.1g)、LiOAc(0.4g)、70%(体积分数) 乙酸水溶液(3.0mL)、3.5MPa O2、140℃、15h条件下反应,反应结束后采用依利特3100型高效液相色谱仪对反应液进行分析。
1.7 动力学
动力学参照文献[20]的研究方法,具体如下:在最优的反应条件下,分别以苯和苯酚作为反应底物,分别获得苯和苯酚浓度随时间变化的动力学曲线,利用一级反应的动力学方程-ln(1-X)=kt对反应进行判断(式中,X为转化率,k为速率常数,t为反应时间),确定苯和苯酚的氧化均为一级反应,并获得相应的反应速率常数。
2 结果与讨论
2.1 催化剂表征
2.1.1 扫描电子显微镜分析
采用扫描电子显微镜(SEM)观察了催化剂的表面形貌。如图1 所示,HPG 和HCG 显示出较小的颗粒结构,表明催化剂前体的改变对其表面形貌无明显影响。此外,与使用前的HCG 相比,反应后的催化剂形貌并无明显变化。

图1 HPG、HCG和使用后HCG催化剂的SEM图像
2.1.2 XRD分析
催化剂的XRD谱图如图2所示。所有催化剂均在2θ=25°附近观察到一个碳的(002)晶面特征峰,表明催化剂主要由无定形碳构成[21]。此外,不同的醛类前体对催化剂的晶型影响不大,且使用后的催化剂晶型未发生明显改变。
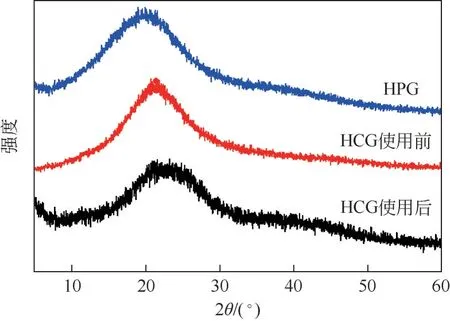
图2 HPG、HCG和使用后HCG催化剂的XRD谱图
2.1.3 FTIR分析
利用FTIR 光谱对催化剂的官能团进行表征,结果如图3 所示。所有样品在3300~3600cm-1处的峰归因于—OH 的特征吸收峰[22],在2858cm-1和2930cm-1处的特征吸收峰分别归因于C—H 键和—CH2—中C—H 键的伸缩振动[23]。1600cm-1和1700cm-1处的峰归因于C= = C双键和羰基中C= = O键的特征吸收峰[22,24]。在1430~1460cm-1处的峰归因于聚合物分子中亚甲基连接键中σC-H的特征吸收峰[25],在1370cm-1处的特征吸收峰归因于—OH 的平面拉伸[26]。与HPG 相比,HCG 在1600cm-1处的C= = C键的吸收峰和1700cm-1处的C= = O键的吸收峰的相对强度均有所增强,这表明HCG 较HPG 样品中C= = C 键和C= = O 键的含量更高。对HCG 使用前后的红外谱图进行对比,结果显示,所有的出峰位置和主要峰型都未发生变化,表明催化剂在使用后的主体结构没有发生变化。但是使用后催化剂在1600cm-1处的羰基C= = C 键的吸收峰的强度相对增强,表明使用后的HCG 中C= = C 键含量更高;在2858cm-1和2930cm-1处的吸收峰强度也有明显的减弱,表明使用后的HCG 催化剂中的C—H 键和—CH2—含量相对减少;在1700cm-1处的吸收峰强度略微减少,表明使用后的HCG 催化剂中的C= = O相对含量略微降低。
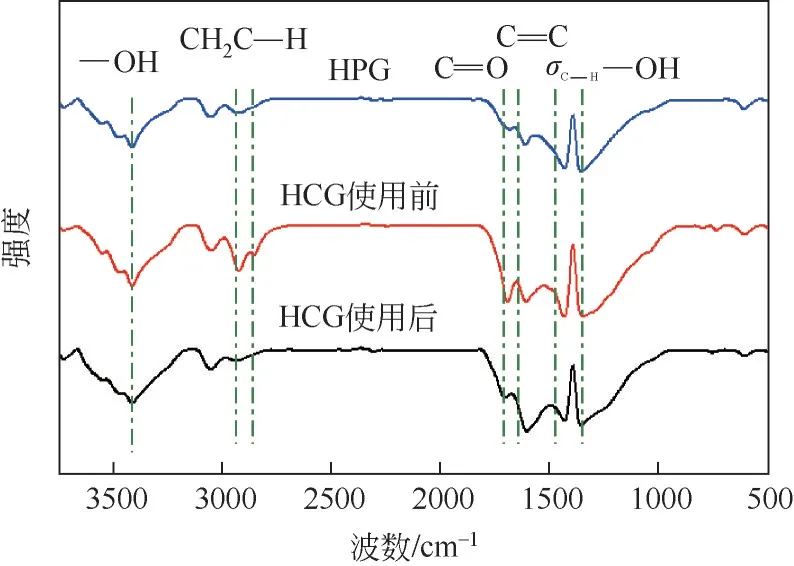
图3 HPG、HCG和使用后HCG催化剂的FTIR谱图
2.1.4 N2吸附-脱附
通过N2吸附-脱附测定了催化剂的孔结构和类型(表1)。结果显示,所有样品的比表面积介于7.4~9.4m2/g 之间,孔体积介于0.0086~0.0148cm3/g之间。此外,平均孔径介于4.7~6.6nm 之间,表明该系列材料具有介孔结构[27]。与新鲜催化剂相比,使用后的催化剂比表面积和孔径均有所增大,这可能是由于催化剂在高温有机溶剂条件下剥层导致。
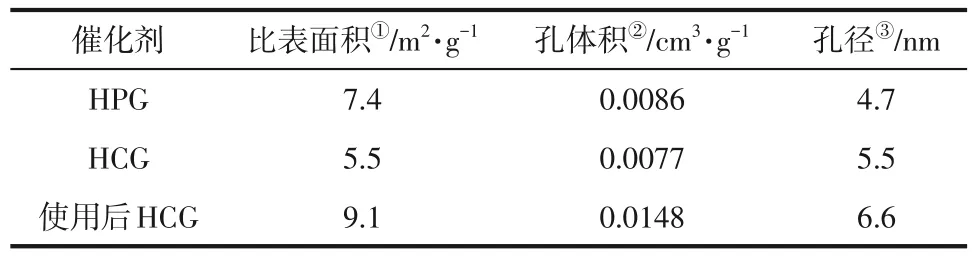
表1 HPG、HCG和使用后HCG催化剂的结构特性
2.1.5 XPS分析
使用XPS光电子能谱测定了催化剂表面元素的化学键合状态,结果如图4 所示。在图4(a)中,HCG 的C 1s 能谱在284.8eV、286.6eV 和288.7eV 处显示出三个特征峰,其分别归属于C= = C/C—C、C—O 和C= = O 基 团[28-30]。 如 图4(b) 所 示, 在532.1eV、533.3eV 和536.2eV 处出现三个峰,分别归属于酮羰基C= = O基团、羟基C—OH基团和表面吸附的水分子[31-33]。结合图4 测试结果,与新鲜催化剂相比,使用后催化剂表面的酮C= = O 键含量增加,而表面的羟基含量降低,这可能是由于反应过程中,催化剂表面部分C—OH 被氧气氧化为酮C= = O[34]。该结论与红外中观察到的HCG 使用后催化剂中C= = O 减少的现象不一致。实际上,XPS 只能探测催化剂表面10nm 以内厚度的信息,而红外检测的是物质的整体结构信息。因此,可以推断HCG使用后催化剂的C= = O总含量略有降低,但是其表面的C= = O含量有所增加。
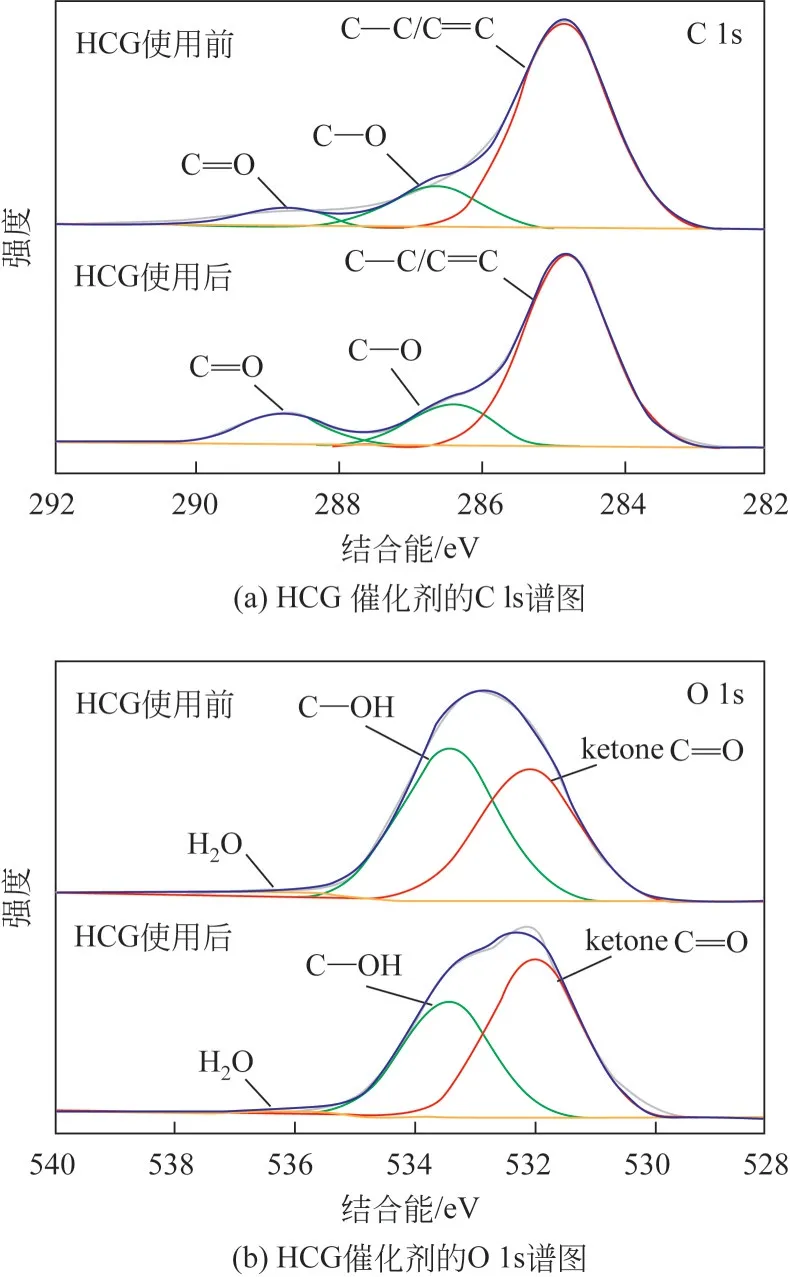
图4 新鲜和使用后HCG催化剂XPS光谱
2.2 催化剂性能
考察了不同催化剂对氧气氧化苯制备苯酚的催化性能的影响,结果如表2所示。在不加入催化剂或无氧气的条件下,体系中并没有检测到苯酚的生成,表明催化剂和分子氧在反应中起着至关重要的作用(表2,序号1、2)。此外,在无添加剂LiOAc的条件下,苯酚产率仅4.8%,表明添加剂对其产率有着较大影响(表2,序号3)。通过改变酮前体所制备的催化剂在相同条件下均表现出相似的催化性能(表2,序号4、5),其中以环己酮和乙二醛为前体,通过简单水热法制备的催化剂的催化性能较好,苯酚产率达到12.5%。

表2 不同催化剂对苯羟基化制苯酚的催化性能①
以HCG 为催化剂,考察了反应条件(溶剂、反应温度、反应时间、催化剂用量、氧气压力和添加剂用量等)对反应性能的影响,结果如图5 所示。探究了不同溶剂对反应性能的影响[图5(a)],当使用乙酸水溶液为溶剂时,苯酚收率为16.3%。当以其他有机试剂作为反应溶剂时,反应得到微量苯酚或没有苯酚生成。因此,选择乙酸水溶液作为后续实验的反应溶剂。温度通常对反应性能的影响较大[图5(b)],当反应温度从110℃升高到150℃时,苯酚的收率逐渐增加;当温度高于140℃时,苯酚的收率开始降低。这是由于苯酚在较高温度下向生成对苯二酚、对苯醌、二氧化碳等副产物转化。随着反应时间的延长,苯酚的收率明显增加[图5(c)]。但当反应时间超过15h 后,苯酚的收率逐渐降低,表明苯酚被进一步氧化为对苯二酚、醌等副产物。此外,催化剂的用量对反应也有着重要影响[图5(d)]。随着催化剂用量的增加,苯酚的收率逐渐增加,这是由于增加活性位点提高了反应速率。当催化剂过量时,苯酚的收率大幅降低,这是由于过量的活性位点使得苯酚未发生及时脱附,而被进一步氧化成为相应的副产物。氧气的压力也会影响反应的活性,随着氧气压力从2.0MPa 升高到4.0MPa,苯酚的收率呈现出先升高后降低的趋势。这是由于氧气压力增大会使得溶剂中溶解的氧增多,从而提高反应活性;但是过多的氧气会使得反应中产生的活性氧含量不断增加而使苯酚过度氧化生成二酚和醌等副产物。因此,氧气压力在3.5MPa 时苯酚产率最佳[图5(e)]。氧气氧化苯制备苯酚反应体系中,LiOAc 的加入会提高苯酚的收率。当LiOAc 的用量从0.2g 增加到0.6g 时,苯酚的产率先升高后降低[图5(f)],可能是过多的LiOAc抑制了氧自由基的产生[9],苯酚的产率降低。因此得出最优反应条件为:5mmol 苯,100mg 催化剂,3mL 70%(体积分数)乙酸水溶液,0.4gLiOAc,140℃,15h。在最优条件下,苯酚产率为16.3%。
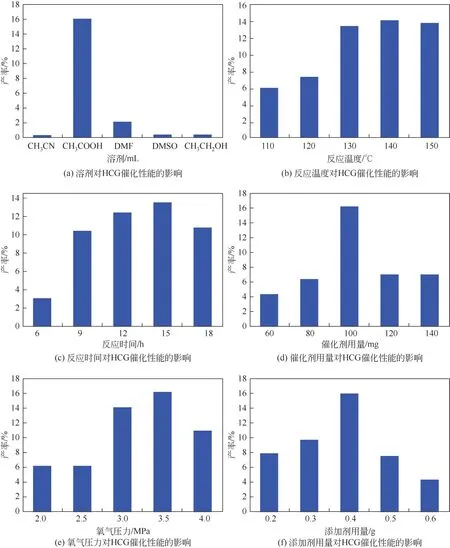
图5 不同因素对HCG催化性能的影响
2.3 HCG催化反应动力学
为了进一步研究该反应,对苯的连续氧化反应(图6)进行了动力学研究。
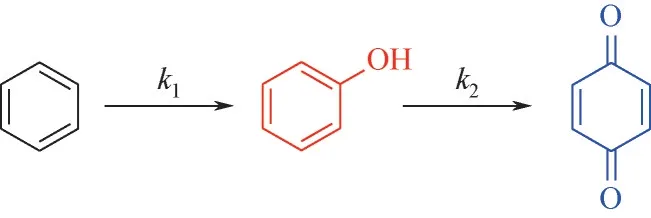
图6 苯连续氧化反应示意图
首先,通过动力学曲线确定了连续反应中的苯氧化到苯酚和苯酚到苯醌的反应级数。以-ln(1-C)对时间进行作图,其中C为转化率,如图7 所示。从图中可以看出,苯氧化和苯酚的氧化反应中-ln(1-C)和时间均具有良好的线性关系,表明这个连续反应的两步均为一级反应。苯氧化生成苯酚和苯酚氧化生成苯醌的速率常数k1和k2值分别为0.02335h-1和0.11578h-1。从反应速率常数可以看出该反应体系中苯酚在动力学上比苯更容易发生氧化。
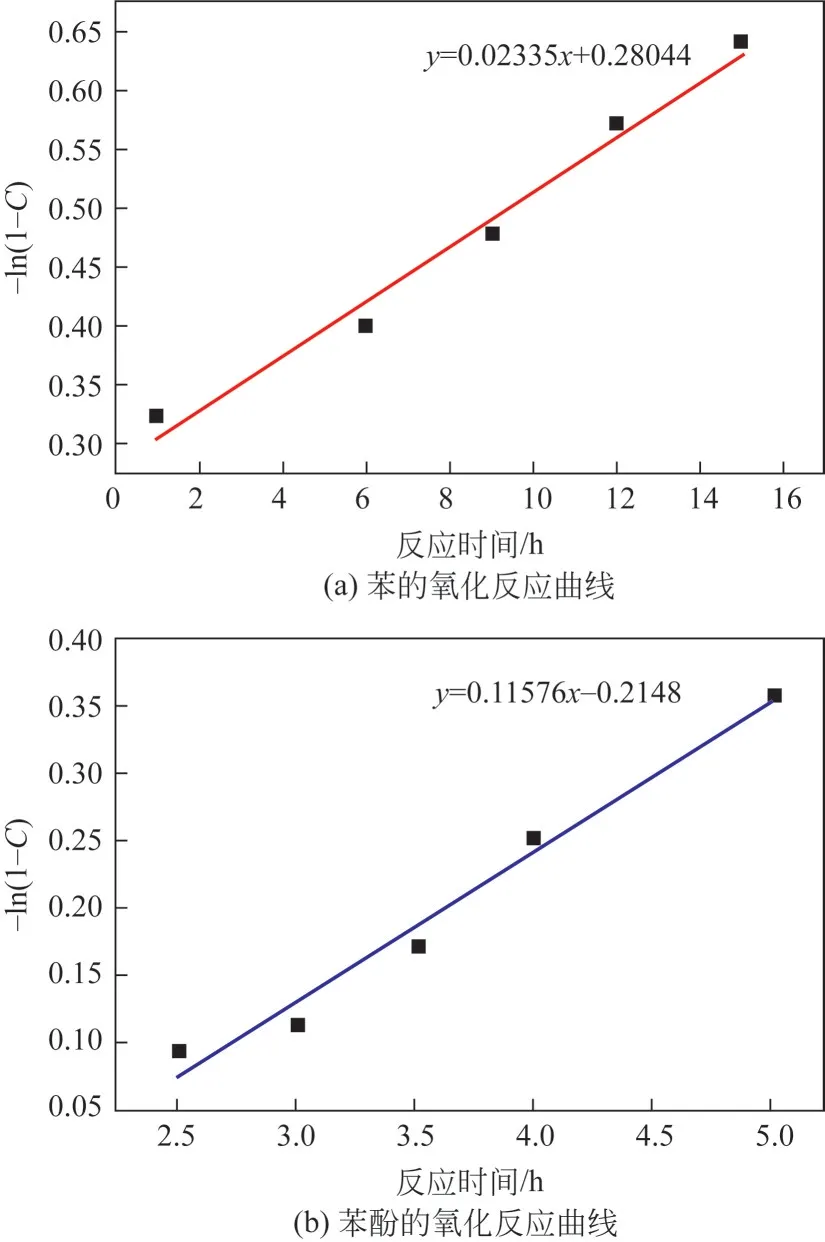
图7 不同底物的氧化反应曲线
为了进一步研究HCG催化剂对连续反应性能的影响,利用阿伦尼乌斯公式计算了苯氧化到苯酚和苯酚氧化到苯醌的表观活化能(Ea)。如图8所示,苯氧化到苯酚的表观活化能(Ea)为48.2kJ/mol。该反应表观活化能与H2O2氧化苯制备苯酚的活化能(43~57kJ/mol)基本一致[35-38]。特别是该表观活化能与化学处理的石墨烯催化H2O2氧化苯制备苯酚的表观活化能(45kJ/mol)一致[34],也与文献报道的羟基自由基·OH 氧化苯制备苯酚的活化能[(50.3±2.5)kJ/mol]完全一致[36]。这些表明本文中的非金属催化剂催化反应机理与H2O2氧化苯制备苯酚的机理类似;同时,该反应的决速步应该是羟基自由基·OH 进攻苯环的反应。另外,苯酚氧化到苯醌的表观活化能为25.4kJ/mol。这表明苯酚在动力学上比苯更易发生氧化反应。因此,后续研究需要从动力学的角度出发,设计提高苯氧化的反应速率,降低苯酚的氧化反应速率。
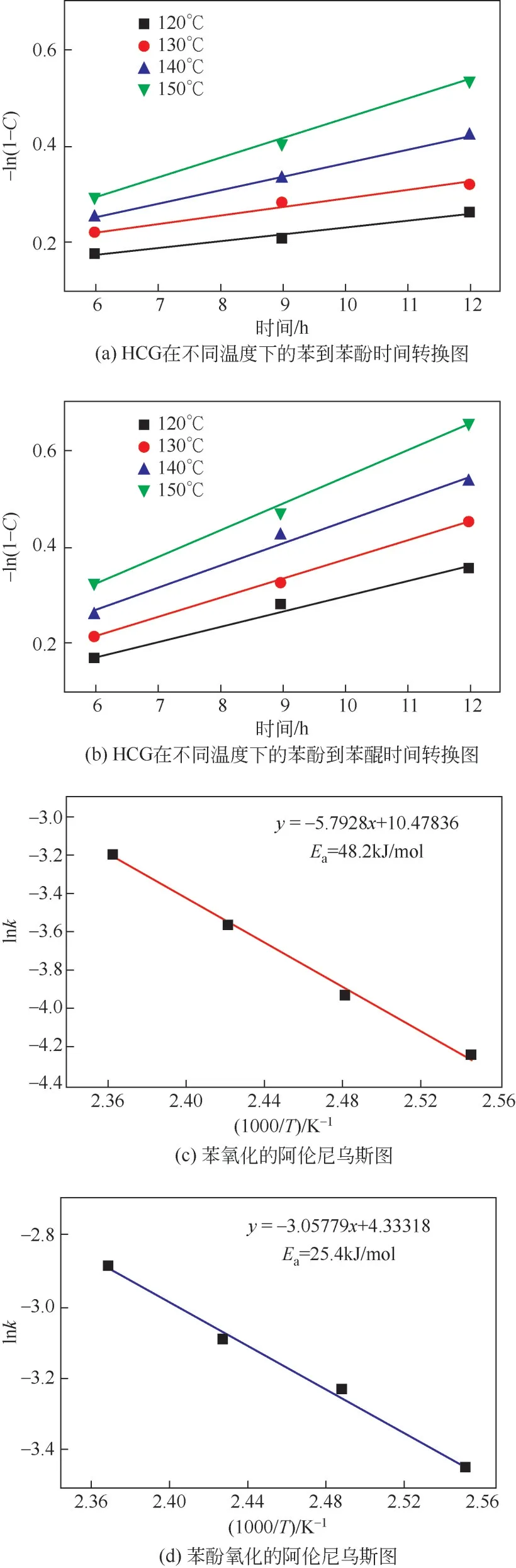
图8 苯和苯酚氧化反应的动力学①
2.4 反应机理
为了探明HCG 催化剂活性位点,以环己酮、乙二醛和环己醇分别作为小分子催化剂催化苯制备苯酚(表3,序号1~3)。使用环己酮作为催化剂催化反应时,苯酚产率为13.3%,而以乙二醛和环己醇为催化剂的苯酚产率仅不足1%。表明在HCG催化剂中,环酮C= = O 基团为主要活性位点。为了进一步探明反应是否为自由基反应,在相同条件中加入自由基清除剂叔丁醇(TBA,·OH清除剂)和2,6-二叔丁基对甲酚(BHT,·O-2清除剂),苯酚的产率分别降至3.7%和1.6%(表3,序号4~7),表明羟基自由基(·OH)和超氧自由基(·O-2)都参与了反应。此外,在相同的反应条件但没有氧气的条件下(N2气氛)进行反应,发现没有苯酚生成(表4,序号8),这表明生成的苯酚中的氧原子来自于O2,而非来自于水分子或催化剂表面含氧基团。当将氧化剂换成H2O2时,有6.3%的苯酚生成,这表明H2O2能够在催化剂的进一步作用下羟基化苯生成苯酚。为研究添加剂在反应体系中的作用,使用LiCl 代替LiOAc 添加剂(表3,序号9),苯酚产率为1.7%;使用NaOAc 代替LiOAc 时,苯酚产率为12.5%(表3,序号10)。这表明Li+在添加剂中起的不是主要作用,OAc-发挥主要作用,这是由于OAc-和乙酸水溶液形成的缓冲溶液提供了酸性环境[39]。

表3 苯羟基化制苯酚的对照实验①
结合文献报道[15-16,40]和前面的实验,推测HCG催化剂催化氧气氧化苯制备苯酚可能的催化机理如图9 所示。在酸性环境下环酮C= = O 与H+作用形成烯醇式[41],烯醇式在氧气气氛下与O2反应并生成超氧自由基·O-2[42-43];在酸性环境下超氧自由基·O-2与H+生成H2O2;H2O2在催化剂的作用下进一步生成羟基自由基·OH[44];羟基自由基·OH 进攻苯环上的C—H键生成苯酚[8]。
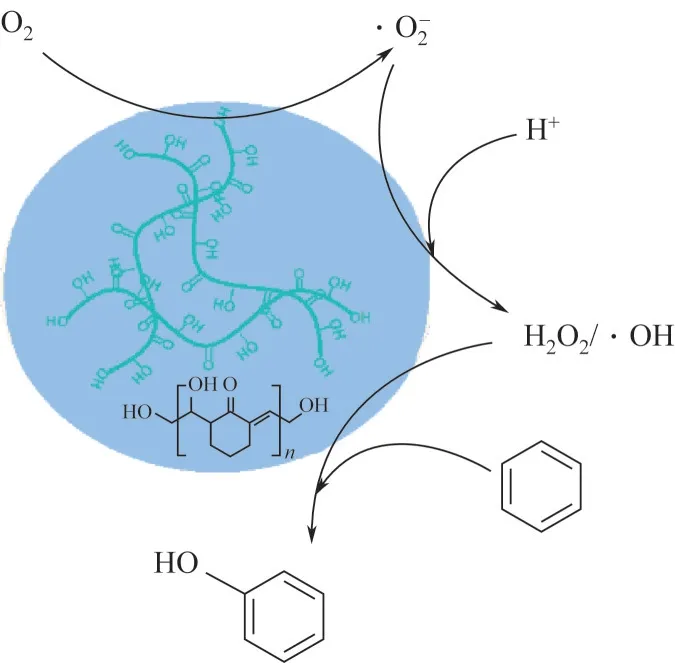
图9 HCG催化苯羟基化制备苯酚的反应机理
2.5 循环实验和热过滤实验
为了测试HCG 催化剂的稳定性,进行了热过滤实验[图10(a)]。在反应9h后离心分离催化剂,然后将反应液在相同条件下继续反应6h,反应液中苯酚的产率并没有提高,表明反应过程是遵循非均相反应进行的。通过循环实验对催化剂的可重复使用性进行测试。如图10(b)所示,经过三次循环后,苯酚的产率由16.3%降至4.8%,表明催化剂的活性逐渐降低。通过收集反应后的催化剂发现,使用后的催化剂质量小于加入的新鲜催化剂的量,表明在反应过程中有少量的催化剂流失。除此之外,在不添加底物苯、其他条件均相同的情况下进行对照反应,反应结束后通过气相色谱仪和气质联用仪检测出反应混合液中含有少量环己酮和乙二醛。这表明催化剂中部分结构在反应过程中发生解聚,从而失去了部分活性位点。同时,使用后催化剂中的C—H 键和—CH2—含量相对减少(图3),进一步证明了催化剂发生部分解聚。
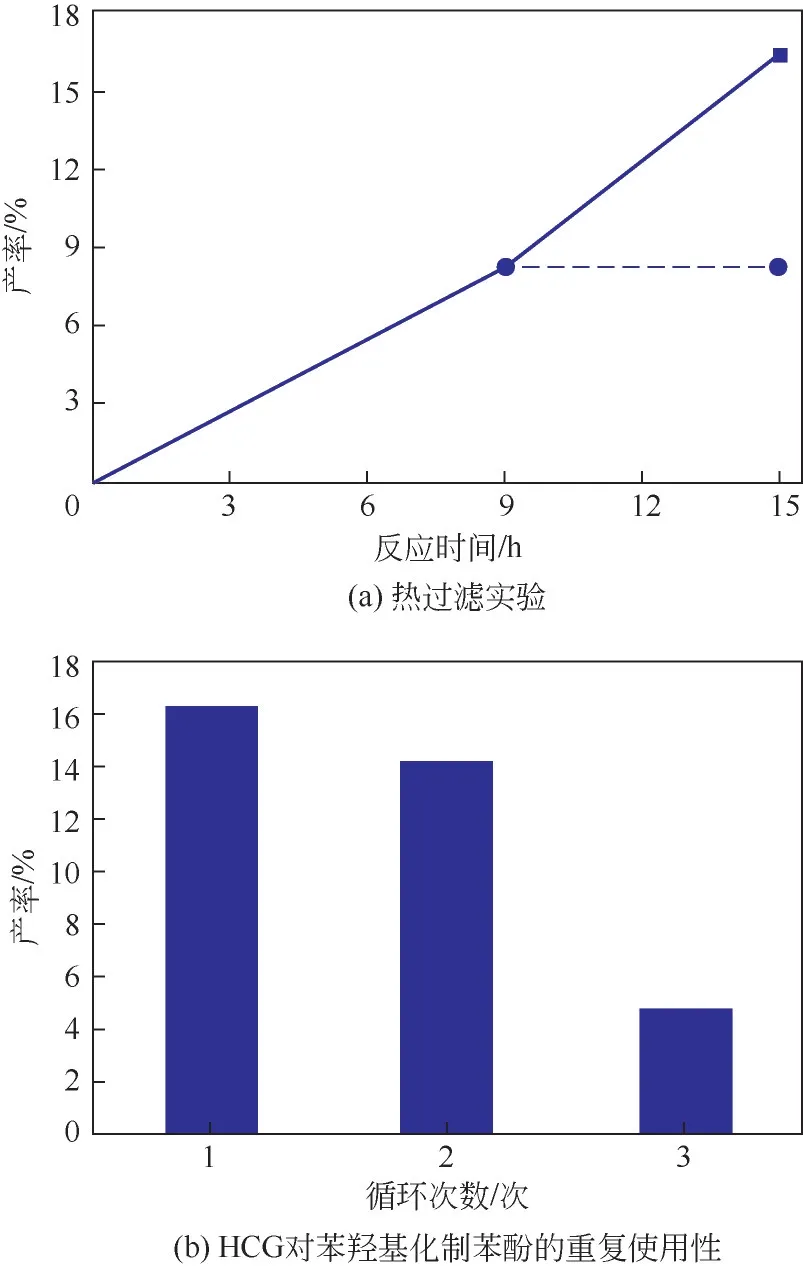
图10 HCG稳定性研究结果
3 结论
通过简单水热法制备了改性的醛酮聚合物,其可作为非金属催化剂催化氧气氧化苯制备苯酚。通过XRD、FTIR、XPS 等表征手段证明了制备的HCG 催化剂为无定形碳结构且表面含有丰富的环酮C= = O基团。其中,环酮C= = O基团是有效催化苯与氧气羟基化的关键活性位点。催化苯氧化实验表明,HCG 催化剂表现出较高的催化活性,苯酚的最佳产率可以达到16.3%。此外,通过自由基捕获及对比实验证明了反应机理为自由基反应。这为设计苯羟基化制苯酚的非金属催化剂提供了一条新思路。
猜你喜欢
云南化工(2020年11期)2021-01-14
云南化工(2020年11期)2021-01-14
应用化工(2020年9期)2020-09-29
中成药(2018年2期)2018-05-09
新乡学院学报(2016年6期)2016-12-01
当代化工研究(2016年9期)2016-03-20
合成化学(2015年4期)2016-01-17
中南民族大学学报(自然科学版)(2014年4期)2014-08-06
化工生产与技术(2014年6期)2014-02-27
化工生产与技术(2014年6期)2014-02-27
