
X连锁隐性鱼鳞病基因型表型分析
2023-10-13 03:40:30崔梦醒杨逸飞黄鹤群
中国麻风皮肤病杂志 2023年10期
陆 鹏 陈 刚 崔梦醒 杨逸飞 梁 骜 黄鹤群 梁 波
1安徽医科大学第一附属医院皮肤性病科,安徽合肥,230022;2安徽医科大学第一附属医院检验科,安徽合肥,230022;3安徽医科大学第一临床医学院,安徽合肥,230022
X连锁隐性遗传性鱼鳞病(OMIM 308100,X-linked ichthyosis,XLI),也被称为类固醇硫酸酯酶(steroid sulfatase,STS)缺乏症,主要由STS催化功能缺失引发[1]。XLI是遗传性鱼鳞病的第二大常见形式,患病率仅次于寻常型鱼鳞病,全球发病率为1∶1500至1∶6000[2],好发于男性(女性多为无症状携带者),症状常起始于新生儿或出生后不久[3]。典型的临床表现为皮屑脱落,皮肤表面干燥,躯干部位及四肢多边形褐色鳞片,病情常冬重夏轻。由于相近的临床皮损,XLI可与寻常型鱼鳞病、银屑病、表皮松解性鱼鳞病等脱屑性皮肤病相混淆,因而,容易发生误诊或漏诊[4]。
本研究收集4例汉族XLI患者外周血及临床信息,采用全外显子组测序技术检测基因突变情况,同时采用拷贝数变异(copy number variation, CNV)分析及目标区域琼脂糖凝胶电泳进行验证。对FLG突变位点进行Sanger 测序验证。
1 资料和方法
1.1 临床资料 4例汉族男性XLI病例为安徽医科大学第一附属医院皮肤科门诊就诊患者。除皮肤表现外无其他器官异常。患者1来自安徽省,27岁,出生时眉弓上方及躯干轻微鳞屑,随年龄增长皮损增加,皮损主要集中于腰部及四肢,可见褐色脱屑,患者有一舅舅有类似症状。患者2来自安徽省,41岁,出生后四肢干燥,后泛发全身,胫前伴有抓痕,患者有一外甥有类似症状。患者3来自河北省,31岁,出生后三个月腹部出现鱼鳞样脱屑,后逐渐向四肢蔓延,可见四肢干皮样脱屑,患者曾被诊断为寻常型鱼鳞病,家中有一舅舅有类似症状。患者4来自安徽省,18岁,出生后脚背有轻微脱屑,后泛发全身,冬季加重夏季缓解,现全身可见褐色多边形鳞片样皮损,并伴有黑褐色角化颗粒,病症尤以膝关节和肘关节为重,该患者曾被诊断为表皮松解性鱼鳞病,家族史不详。患者1~3家系图见图1。患者临床照片见图2。
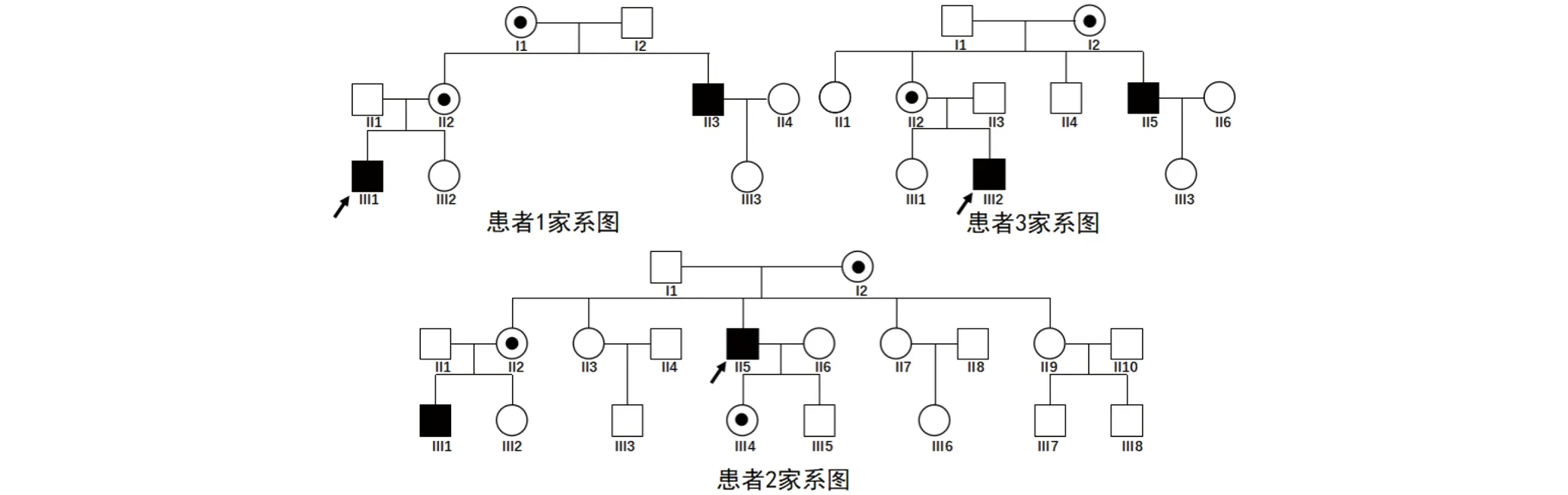
图1 隐性X连锁鱼鳞病患者(1~3)家系图,黑圆点为携带者,黑色方形为XLI患者

图2 4例隐性X连锁鱼鳞病患者的临床表现,下肢可见褐色斑块形鱼鳞样皮损(2a~2d分别对应患者1~4)
1.2 方法
1.2.1 DNA提取 本研究得到安徽医科大学第一附属医院伦理会批准,并按照《赫尔辛基宣言》执行,获取入组4例患者签署的知情同意书,然后填写调查表信息并采集典型临床照片,同时取4 mL外周静脉血,采用FlexiGene DNA试剂盒(Qiagen,德国)抽提基因组DNA。
1.2.2 全外显子组测序 构建DNA文库后用探针将全部外显子区域富集,再将外显子文库进行Illumina Hiseq2000平台高通量测序。将测序得到的bcl文件用CASAVA1.8软件转换为fastq文件,再将全部测序读长用BWA、Samtools和Picard软件比对到人类参考基因组(版本为GRCh37/hg19)上,然后用GATK系列软件将生成的bam文件进行局部重新比对,去掉重复序列后利用其中的Haplotypecaller进行变异检测并导出vcf变异文件,最后用Annovar软件对vcf变异文件进行注释。另外,通过计算各探针捕获区域的相对平均测序深度分析CNV。
1.2.3 琼脂糖凝胶电泳 根据STS基因参考序列设计引物,对患者及健康对照的STS基因外显子进行聚合酶链反应(Polymerase chain reaction,PCR)扩增,扩增产物采用2%琼脂糖凝胶电泳分析。引物序列及扩增条件见表1~3。

表1 STS基因2-11号外显子PCR扩增引物序列

表2 PCR体系

表3 PCR条件
1.2.4 突变分析及Sanger测序 突变通过人群频率进行筛选,查询ClinVar数据库突变的致病性,通过scsnv、revel、Func.refGene等工具搜寻可疑突变,分析拷贝数变异。
根据外显子测序提示FLG突变位点c.5368C>T设计引物,上游引物:CGGGAGACATCAGACCTTTC,下游引物:CGAGGGTCCAGTGGTAGCCA,进行Sanger测序验证。
2 结果
2.1 STS基因检测 CNV分析显示患者STS基因的外显子区域基本无读长覆盖(见图3)。患者相对平均测序深度(relative average sequencing depth, RASD)在0.4~0.6的探针覆盖区域标记为蓝色,相对平均测序深度小于0.1的探针覆盖区域标为红色。和蓝色区域相比,红色区域的相对平均测序深度基本为0,这表明红色区域缺失。患者母亲CNV分析显示,和蓝色区域相比,红色区域相对平均测序深度降低了大约一半,这表明红色区域存在杂合缺失。
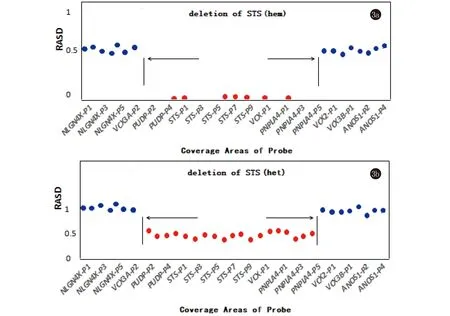
图3 STS基因外显子拷贝数变异分析(3a:患者:3b:患者母亲)
PCR扩增产物琼脂糖凝胶电泳显示4例患者STS基因10个外显子无相应扩增产物,健康对照扩增出相应大小扩增片段。见图4。
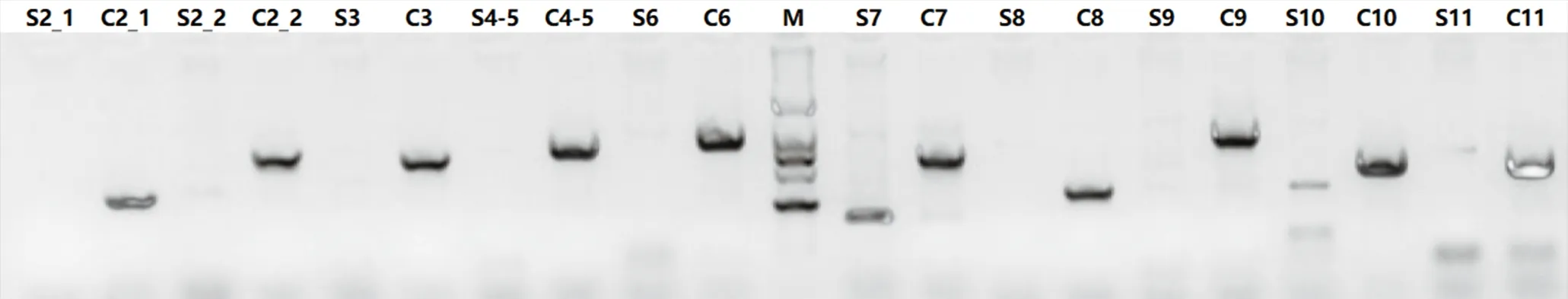
图4 PCR扩增产物琼脂糖凝胶电泳图 S2-S11:患者;C2-C11:对照
2.2 FLG基因检测 外显子测序显示患者1~3未发现FLG基因异常,4号患者携带一个已报道的FLG基因无义杂合突变:c.5368C>T(p.Gln1790Ter),在4号患者及健康对照中Sanger测序证实。见图5。
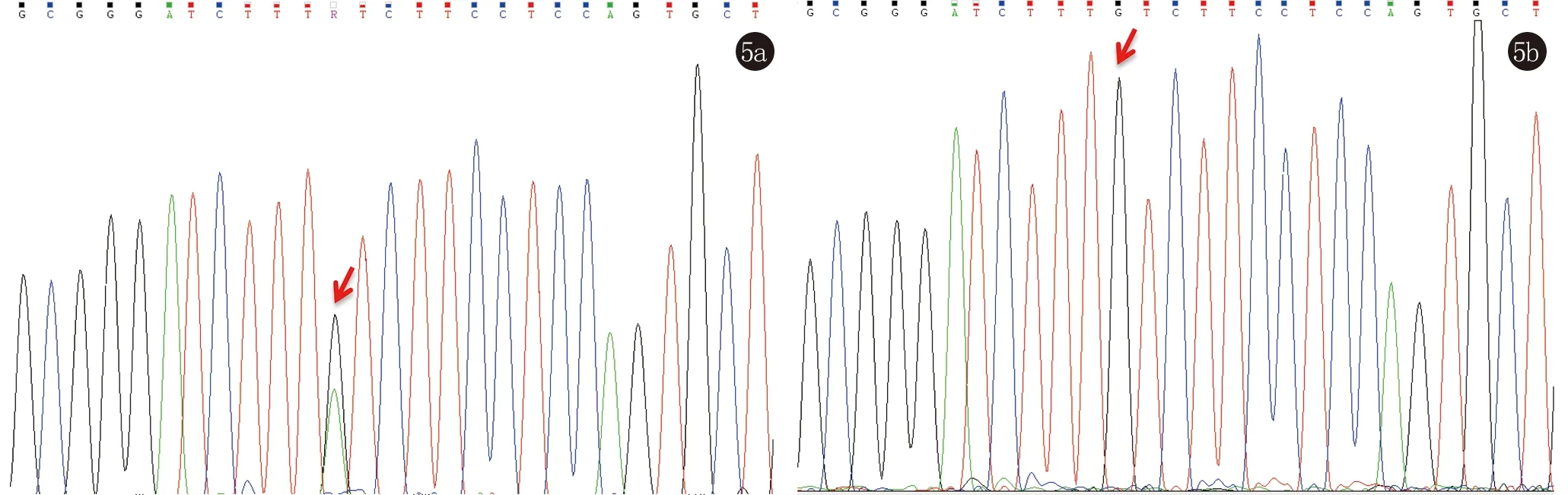
5a:患者4杂合突变;5b:对照野生型图5 FLG c.5368C>T突变Sanger测序图
3 讨论
遗传性鱼鳞病是一组先天性鱼鳞病,病变不只局限于皮肤,其遗传模式包括性连锁隐性遗传,常染色体显性遗传,常染色体半显性遗传,常染色体隐性遗传等。XLI为常见遗传性鱼鳞病,除皮肤鳞屑外,也可累及身体其他系统和器官,如出现角膜混浊、智力障碍、隐睾等症状,危害较大[5]。本研究的4例患者仅表现皮肤症状,但表型明显存在差异。其中,患者3症状轻微,主要表现为胫前鳞状皮损,余部仅表现为干皮样脱屑。患者4皮损严重程度最高,膝盖和关节部位可见污垢样皮损,全身均覆盖有褐色多边形皮损。XLI与其他脱屑性疾病有相似的临床表现,对于不典型的XLI患者,临床上容易发生误诊或漏诊,可能被诊断为寻常型鱼鳞病或角化型鱼鳞病。尤为值得注意的是,严重型寻常型鱼鳞病和轻微型XLI的表型存在交叉重叠,仅根据患者的临床特征难以做出准确诊断。本研究4例患者,有2例曾被诊断为其他疾病。因此,对性连锁隐性鱼鳞病病史的采集至关重要,同时,基因检测对可疑病例的确诊不可或缺[6]。
全球范围内,约90%XLI病例由STS基因完全缺失所致,少数患者可见STS基因部分缺失及点突变。纳入本研究的4例患者均为STS基因完全缺失,与既往报道一致,这提示STS基因完全缺失可能也是我国XLI发病的主要原因。STS基因是循环胆固醇利用的关键基因,含有10个外显子,编码一种定位于内质网的多通道膜蛋白。STS属于硫酸酯酶家族,水解3-β-羟基类固醇硫酸盐,作为雌激素,雄激素和胆固醇的代谢前提。STS功能的完整性与皮肤及其附属器的发育和功能维持紧密相关。
此外,本研究中1例患者(患者4)出现FLG提前终止突变,c.5368C>T (p.Gln1790Ter)。该突变为已知突变,曾在多个人群有过报道[7]。生物信息学工具SIFT及MutationTaster均预测突变效应为“Damage”,提示为致病突变,人类孟德尔遗传在线网站(OMIM)显示该突变与特应性皮炎及寻常型鱼鳞病相关。gnomAD数据库显示c.5368C>T最小等位基因频率为0.00004467,提示该突变在正常人群有一定的携带率。既往XLI患者队列中进行FLG突变研究主要在欧洲国家,丹麦的一项109例XLI患者队列中,有66例患者做了FLG突变检测,其中23例发现了FLG突变(34.85%)[4],德国和澳大利亚的一项51例XLI患者队列中,FLG突变的患者是9例(17.6%)[8],我们的研究中4例患者中有1例出现FLG突变(25%),XLI患者中FLG突变的频率显著高于正常对照人群,这提示FLG突变可能也是汉族XLI患者中一个常见的修饰因子。FLG突变与XLI的临床严重程度也存在关联,在苏格兰的一个家族中,同父异母的哥哥和弟弟都是XLI患者,哥哥伴有FLG突变(p.Arg501Ter),表现出了类似表皮松解性鱼鳞病的更严重表型[9]。中国的一个XLS家系中STS和FLG突变(p.Ser1107SerfsTer15)的突变加重了IV表型,其他地区的病例也支持了STS和FLG突变的共同作用可能加重皮损表现的理论[10,11]。我们研究中,带有FLG突变的患者表现出了更为严重的表型,膝关节有密集黑褐色角化颗粒,和既往报道结论一致。
从功能上来说,STS催化活性的缺失导致胆固醇硫酸盐水解减少,从而积聚角质层,影响角质层脂类组分[12]。FLG基因提前终止突变导致编码的丝聚蛋白的截短和蛋白功能的不完整性[13],进而影响角质层中的蛋白质组分,使得角质层中的脂类组分和蛋白质组分均发生变化,从而加重皮肤屏障的损害[14]。皮肤屏障受损使皮肤防御功能下降,引发感染和炎症反应,从而进一步加剧遗传性鱼鳞病的临床表型[15]。
本研究扩展了对患者的基因型和表型的认识,FLG基因与XLI表型间的关系有待进一步研究阐明。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29 01:57:52
中国临床保健杂志(2021年3期)2021-04-03 14:38:13
——以安徽医科大学为例
内蒙古科技与经济(2021年4期)2021-03-26 09:14:18
中国生殖健康(2020年4期)2021-01-18 02:58:10
中国生殖健康(2018年4期)2018-11-06 07:12:16
安徽医科大学学报(2016年12期)2017-01-15 14:22:02
China Report Asean(2016年2期)2016-09-26 03:23:23
中华皮肤科杂志(2014年4期)2014-12-19 12:55:46
中国民族民间医药·上半月(2014年2期)2014-11-27 21:13:55
湖北农业科学(2014年11期)2014-09-10 18:06:07
