
分子动力学在耐火材料研究中的应用进展
2023-10-10 06:19刘德嵩罗旭东
辽宁科技学院学报 2023年4期
祁 欣,刘德嵩,罗旭东,张 磊
(1.辽宁科技学院 冶金与材料工程学院,辽宁 本溪 117004;2.安徽海螺暹罗耐火材料有限公司,安徽 芜湖 241070)
0 引言
耐火材料是以铝矾土、硅石、菱镁矿、白云石等天然矿石、某些工业原料和人工合成莫来石、尖晶石、碳化硅等为原料,经加工后制造的无机非金属材料,是用作高温窑炉等热工设备的结构材料。
分子动力学研究耐火材料,是对材料的性能和特点提供关键性的参考依据,也是对理论计算和实验的有力补充。分子动力学对分子大小和形状、与其他分子的相互作用、压力下的行为及一种状态与另一种状态相比的相对频率进行定量预测,对化学、物理、材料及其他领域都是至关重要的。分子动力学能够在原子或分子尺度上进行建模计算,模拟结果与实验结果误差较小,因此在耐火材料的界面结合机制、抗渣性、热学性能的研究方面获得了广泛应用。
1 分子动力学
分子动力学是基于牛顿力学模拟分子运动,对系统中各原子运动状态的一种微观描述[1]。原子运动遵循经典的运动规律,其中最常见的形式是牛顿运动方程。对于由N个原子构成的体系,第i个原子有:
Fi=miai
(1)
(2)
其中:Fi为受力矢量;mi为原子质量;ai为加速度;ri为坐标矢量;t为时间。
分子动力学是用一种统计物理的方法,获取一系列状态集合的手段[2]。分开考虑电子和原子核的运动,其哈密顿(Hamilton)量如下:
H=K+u
(3)
其中:H为哈密顿量;K为总动能;u为总势能。
(4)
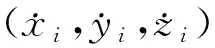
u=u(x1,y1,z1,…,xj,yj,zj,…,xn,yn,zn)
(5)
结合笛卡尔坐标,可以获得系统的运动方程,简称为哈密顿方程组。
(6)
其中:pi为广义坐标矢量;qi为广义动量矢量。
对原子动力学方程组的求解方法包括Euler算法、Verlet算法、蛙跳算法等。其中,Verlet算法形式简单且计算结果准确,性能稳定,普遍应用于众多分子动力学程序中。
分子动力学的基本原理为:建立一个原子系统,根据量子力学计算体系的构型积分,通过对原子动力学方程组进行求解,得到原子空间的运动规律和轨迹,根据物理原理得出该体系的热力学量和其他宏观量,对材料的性能进行理论解释[3]。
2 分子动力学在耐火材料研究中的应用进展
2.1 分子动力学对复相耐火材料界面结合机制的研究
复相耐火材料中异相之间存在界面,界面的结合情况对耐火材料的整体性能有很大影响。复相耐火材料界面位于异相之间,连接界面两侧的物相,传递应力和温度等,构成复相耐火材料整体,复相界面结构如图1所示。
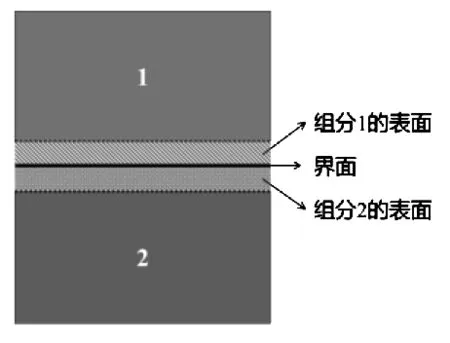
图1 复相界面结构示意图
复相耐火材料界面的分子动力学模拟主要通过Materials Studio软件完成[4]。采用Materials Studio中的Visualizer模块,能够建立各种晶体、无定形及高分子材料的三维结构模型,该模块提供搭建材料结构模型所需要的所有工具,并支持其他模块进行后续处理[5]。通过Materials Studio中的Forcite模块对各界面进行结构优化、分子动力学模拟热处理和冷却过程,分析界面演变过程并计算界面的结合能等参数,建立复相耐火材料异相界面的结合模型。Forcite模块[6]是Materials Studio中经典的分子力学工具,可以对单分子和周期性体系的几何优化、动力学模拟和能量计算,包含COMPASS、Universal等力场。
徐等人[7]利用分子动力学方法研究Si3N4与MgO的界面结合情况,构建两者之间不同位向的界面模型,得到结合强度最高的界面。Gao等人[8]借助分子动力学方法研究Si3N4和石墨之间界面随温度变化的情况,分析界面键的键型、键弛豫及界面结合能等参数。图2为β-Si3N4(1 1 0)//石墨(1 0 0)界面分子动力学模拟前后的截面图。王等人[9]通过分子动力学方法研究界面层对复相耐火材料力学性能的影响,动力学模拟后界面发生弛豫,界面两侧原子向界面中间移动,界面结合强度增加,材料的力学性能也得到提高。李等人[10]对Al2O3和TiC界面的结合能和电子结构进行计算和分析,发现Al2O3(0 0 1)//TiC(1 0 0)界面最稳定,这是因为单层O原子的界面成键效果最好,界面处原子成键时的共价性增强。

图2 β-Si3N4(1 1 0)//石墨(1 0 0)界面分子动力学模拟前后的截面图[8]
分子动力学方法在复相耐火材料界面构型、界面能计算等方面取得了较好的研究成果。近年来以分子动力学方法为代表的原子尺度模拟在国内外受到广泛关注,能够从理论角度更准确地理解界面性能和界面行为,可以为复相耐火材料设计提供指导和参考,将在更深层次上理解和发展复相耐火材料的重要作用。
2.2 分子动力学对耐火材料抗渣性的研究
熔渣是由多种氧化物组成的熔体,具有保温、去杂、回收金属氧化物等重要作用。熔渣及耐火材料在整个冶炼过程中是相互作用的,各种精炼工艺开发以来,精炼渣的研究与开发进展迅速,通过调整渣的成分可以控制钢的质量。抗渣性是耐火材料的一个重要性能,渣可以与耐火材料发生反应从而改变其组成和结构,降低耐火材料的使用寿命,耐火材料熔解到渣中也会改变渣的组成,影响渣对钢水的冶炼作用。采用分子动力学模拟可以弥补实验研究对温度、研究区域等限制问题,熔渣分子动力学模拟过程如图3所示,包括构建初始模型、平衡态计算、信息收集、信息分析等[11]。
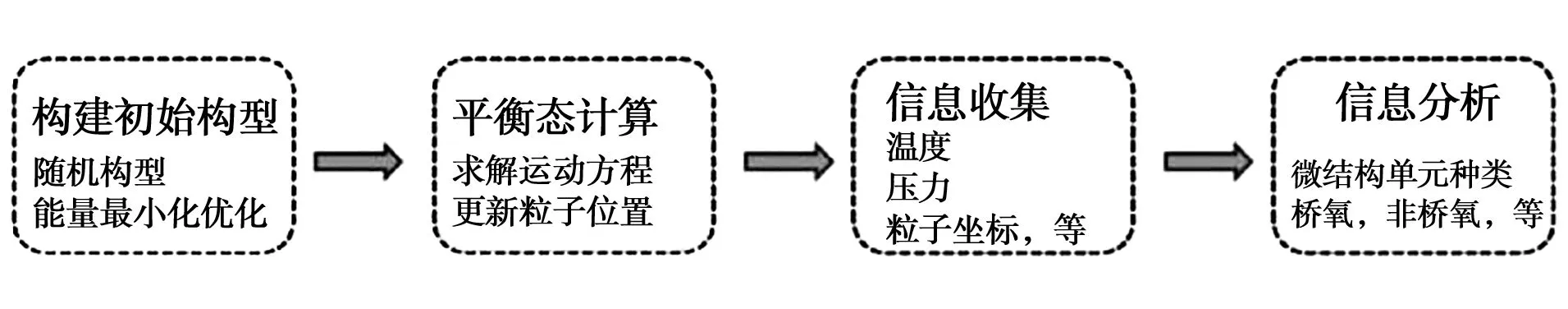
图3 熔渣分子动力学模拟过程[11]
Li等人[12]研究碱度对熔渣中离子的扩散率及黏度的影响,黏度的下降会加重熔渣对耐火材料的侵蚀和渗透,不利于耐火材料的抗渣性。硅酸盐熔渣包括SixOyz-、AlxOyz-等复杂网络结构,如图4所示[11]。周等人[13]通过径向分布函数、均方位移等分析熔渣成分与结构的关系,图5为灰渣和氧化铝分子动力学模拟前后示意图。结构表明,灰渣-Al2O3体系内各粒子的扩散系数相比于灰渣体系增大,其中Mg的扩散系数最大,对Al2O3的渗透能力最强。
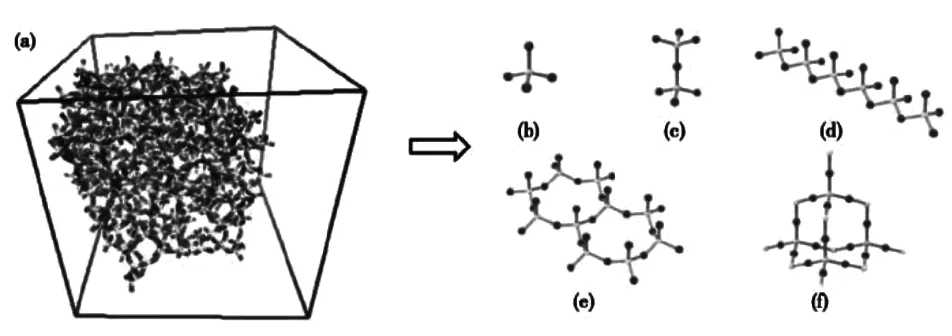
图4 CaO-SiO2-CaF2熔渣的结构示意图:(a)熔体;(b-f)微结构单元[12]

图5 灰渣和氧化铝分子动力学模拟前后示意图[13]
分子动力学模拟作为一个有效的热力学、结构以及传输性质的计算工具,可以从微观的角度直接分析高温熔渣中不同粒子对的键长、键角、配位关系,以及熔渣体系中的氧离子种类和微结构单元种类的分布,还能得到物理模拟实验所不能实现的粒子轨迹图形信息,这些优点使得分子动力学模拟在熔渣的研究中具有显著优势。
2.3 分子动力学对耐火材料热学性能的研究
分子动力学模拟方法与实验方法相比可以精确控制模拟条件,从微观角度解释实验现象,通过模拟预测材料的服役行为,降低了实验周期和经济成本。分子动力学可以用于耐火材料热学性能的计算和分析。
倪等人[14]对钛酸钙和镁铝尖晶石的热导率进行模拟研究,得出材料的导热性能随温度及试样长度变化规律,为实际生产中控制传热提供基础数据,图6为钛酸钙热导率计算模型示意图,图7为温度对钛酸钙热导率影响。Goh W F等人[15]对钛酸锶的热膨胀率等热学性能进行了分子动力学研究,并修正了热导率的有限尺寸效应,模拟结果接近实验数据。

图6 钛酸钙热导率计算模型示意图[14]
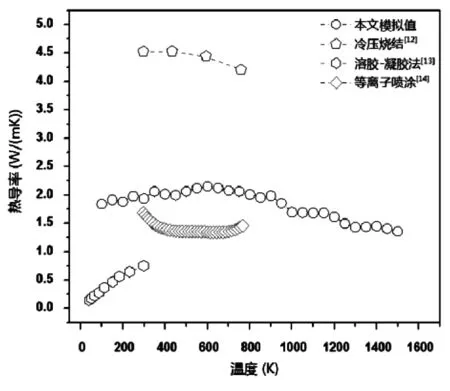
图7 温度对钛酸钙热导率影响[14]
分子动力学对于实验中难以获得的超临界、深过冷等环境都可以实现, 还可以测量实验中无法获得的物理量, 进而从微观角度解释实验现象。 以上可以看出分子动力学在研究耐火材料微纳尺度热学性能方面取得了很好的效果。
3 结语
分子动力学模拟具有计算速度快、模拟与实验结果误差小、研究成本低等优势,在耐火材料的界面结合机制、抗渣性、热学性能等的研究具有可行性,为耐火材料的设计和开发提供了数据支撑。为使分子动力学可以更好地用于耐火材料的研究,提出几点建议:(1)在耐火材料的界面研究中,模拟得到的界面结合情况需要通过TEM等实验手段补充验证,保证后续模拟结果的可靠性;(2)在耐火材料的抗渣性研究中,需要区分不同成网粒子种类带来的影响。即使都是四面体结构,由于键长和键强的不同,同样结构类型的Si-O四面体和Al-O四面体对宏观性能的影响是有区别的;(3)在耐火材料的热学性能研究中,需要明确热学性能的主要影响因素,明确不同计算函数的边界值,提高模拟结果与实验结果的一致性。随着模拟计算科学的逐渐发展及分子动力学在耐火材料研究中的应用,能够从理论角度更准确地理解耐火材料的性能和作用行为,对开发新型高性能耐火材料具有指导意义。
猜你喜欢
中学生数理化·中考版(2023年7期)2023-06-27
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中学生数理化·中考版(2021年4期)2021-07-22
中学生数理化·中考版(2019年4期)2019-05-21
中学生数理化·中考版(2018年3期)2018-05-29
上海金属(2016年2期)2016-11-23
上海金属(2016年3期)2016-11-23
上海金属(2014年2期)2014-12-18
