
Alkyl Chain Engineering of Bithiophene Imide-based Polymer Donor for Organic Solar Cells
2023-10-10 03:29:32BAIYuanqingZHANGJiabinLIUChunchenHUZhichengZHANGKaiHUANGFei
高等学校化学学报 2023年9期
BAI Yuanqing, ZHANG Jiabin, LIU Chunchen, HU Zhicheng, ZHANG Kai, HUANG Fei
Alkyl Chain Engineering of Bithiophene Imide-based Polymer Donor for Organic Solar Cells
BAIYuanqing#, ZHANGJiabin#, LIUChunchen*, HUZhicheng, ZHANGKai, HUANGFei*
(,,,510640,)
Two polymer donor materials, namely pBDT-BTI-EH and pBDT-BTI-ME, were synthesized by copolymerizing benzodithiophene(BDT) unit with bithiophene imide(BTI) unit containing 2-ethylhexyl and methyl alkyl side chains, respectively. Compared to pBDT-BTI-EH∶Y6 based organic solar cells(OSCs), the pBDT-BTI-ME∶Y6-based device exhibited higher charge mobilities, reduced charge recombination, more efficient exciton dissociation, and favorable film morphology, which leaded to increased short current density(sc), fill factor(FF) and thus a significant improvement in power conversion efficiency(PCE) from 9.31% to 15.69%.
Organic solar cell; Polymer donor; Bithiophene imide; Alkyl chain engineering
1 Introduction
As a promising strategy for harnessing clean and renewable solar energy, organic solar cells(OSCs) have garnered significant attention over the past three decades due to the characteristics of lightweight, excellent mechanical flexibility, and potential for roll-to-roll processing at room temperature[1—7]. Recently, the power conversion efficiency(PCE) of OSCs has been improved to 19%[8—13]as a result of the long-term efforts in the innovation of molecular structure[14—18], optimization of processing methods[19—21], and strengthened understanding of device physics[22,23]. One of the critical factors in enhancing the PCE of OSCs lies in the design of novel structures for the donor and acceptor materials in the active layer. Since 2019, Yuan.[24]have synthesized a novel narrow bandgap non-fullerene small molecule acceptor, named Y6, to combine with a wide bandgap(WBG) polymer donor known as PM6 for preparing OSCs with a high PCE of 15.7%. In an effort to further improve the PCE, extensive research has been conducted on Y6, and its derivatives with different end groups[25—27], side chains[28—32], and central electron-withdrawing units[33—36]. However, the choice of WBG polymer donors that exhibit complementary absorption and matching energy levels with Y6 and its derivatives remains limited. In 2020, Ding.[37]developed a novel WBG polymer donor, named D18, which significantly increased the PCE of Y6 acceptor-based OSCs to 18%. These breakthroughs suggest that the development of new WBG polymer donors holds great potential for further improving the PCE of OSCs. Consequently, it is imperative to continue exploring high-performance WBG polymer donors that are well compatible with non-fullerene acceptors.
Efficient WBG polymer donors commonly employ a backbone consisting of alternating electron-donating(D) and electron-accepting(A) building blocks. By adjusting the D and A units, the absorption, optical bandgaps, frontier molecular orbital energies, and hole mobilities of D-A-structrual WBG polymer donors can be easily tuned through the intramolecular charge transfer(ICT) effects and intermolecular D-A interactions[38—41]. Among these polymer donors, benzodithiophene(BDT) has been the most commonly used D unit to construct highly efficient WBG polymer donors such as D18[37], PBDB-T[42], PM6[24], PTzBI[43], PBQ6[44]with various A units, respectively, including 1,3-bis(thiophen-2-yl)-5,7-bis(2-ethylhexyl)benzo-[1,2-c∶ 4,5-c′]dithiophene-4,8-dione(BDD), imide functionalized benzotriazole(TzBI), quinoxaline(Qx). The bithiophene imide(BTI) unit can serve as a promising A building block for constructing organic semiconductors in organic field-effect transistors[45—47], polymer solar cells[48—50], perovskite solar cells[51, 52]and organic thermoelectric[53]due to its planar structure and strong electron-withdrawing capabilities. In addition to modifying the backbone with different D and A units, side chain engineering can also modulate the optoelectronic properties of WBG polymer donors. Adjustable side chains in D units, A units, or-spacers enable the modulation of solubility, absorption spectra, and electronic properties of WBG polymer donors. Moreover, side chain engineering could significantly influence the blend morphology and domain size of the bulk heterojunction[54], thus optimizing the photovoltaic properties of OSCs.
In this study, we synthesized two WBG polymer donors, namely pBDT-BTI-EH and pBDT-BTI-ME, with BDT as the D unit and BTI with varying lengths of side chains as the A unit. Both pBDT-BTI-EH and pBDT-BTI-ME exhibited similar optical and electronic properties. Notably, it was observed that a significant improvement in the PCE of OSCs, from 9.3% to 15.69%, by shorting the alkyl side chains of BTI units from 2-ethylhexyl to methyl. The investigations revealed that this PCE improvement can be mainly attributed to the resulting tighter molecular packing, which enhanced the charge mobility and charge dissociation within active layer. These findings emphasize the importance of rational design and fine-tuning of alkyl side chains in the polymer donor, as they can directly affect the morphology of active layers and eventually play a crucial role in controlling the photovoltaic performance of OSCs.
2 Experimental
2.1 Materials and Measurements
The commercial BDT-based monomers, specifically{4,8-bis[5-(2-ethylhexyl) thiophen-2-yl] benzo [1,2-b∶4,5-b′]dithiophene-2,6-diyl}bis(trimethylstannane)(BDT-DSn) were purchased from Derthon Inc and used as received without further purification. Other chemicals and solvents were purchased from Energy Chemical Co., SunaTech Co., Aladdin Co., J&K Co., etc. and were used without further purification.
The1H NMR and13C NMR spectra were analyzed using a Bruker AV-500 MHz spectrometer. The analysis was conducted using deuterated chloroform(CDCl3) as the solvent at a temperature of 298 K. Tetramethylsiloxane(TMS) was used as the internal standard reference(=0).
The number average molecular weights(n) and polydispersity index(PDI) of copolymers were measured by an Agilent Technologies PL-GPC220 high-temperature chromatograph with polystyrene as the internal standard.
The thermogravimetric(TGA) measurement was performed using NETZSCH TG209F3 equipment under a nitrogen(N2) atmosphere, with a heating rate of 10 ℃/min.
The UV-Vis absorption spectra and temperature-dependent absorption spectra were carried out on a PerkinElmer-Lambda 365 UV-visible spectrophotometer in dilute CB solution, with a temperature range of 20—100 ℃.
Cyclic voltammetry(CV) measurements were performed using a CHI660E electrochemical workstation(Shanghai Chenhua Instrument Company Limited), with a saturated calomel reference electrode, a platinum wire counter electrode, a glass carbon working electrode, and 0.1 mol/L tetrabutylammonium hexafluorophosphate(-Bu4NPF6) as electrolyte.
The surface roughness of the blend film was tested by Bruker Innova atomic force microscope(AFM).
The transmission electron microscopy(TEM) images were obtained by using JEOL JEM⁃2100F field emission electron microscope.
2.2 Synthesis Routes of Monomers and Copolymers
2,2′-Bithiophene-3,3′-dicarboxylic anhydride(compound 1)[55],-(2-ethylhexyl)-2,2′-bithiophene-3,3′- dicarboximide(compound 2a)[56],-(2-ethylhexyl)-5,5′-dibromo-2,2′-bithiophene-3,3′-dicarboximide(compound 3a)[56], trimethyl[4-(2-butyloctyl)thiophen-2-yl]stannane[57]and copolymer pBDT-BTI-ME[58]were synthesized according to the previously reported procedures. Detailed synthesis procedures of monomers and polymers are provided as follows(Scheme 1).

Scheme 1Synthetic routes of copolymers
Synthesis of 2,8-bis[4-(2-butyloctyl)thiophen-2-yl]-5-(2-ethylhexyl)-4H-dithieno[3,2-c:2',3'-e]azepine-4,6(5H)-dione(4a). In a 50 mL two-necked flask, compound 3a(300 mg, 0.6 mmol) and trimethyl[4-(2-butyloctyl)thiophen-2-yl]stannane(748 mg, 1.8 mmol) were dissolved in 20 mL toluene. The solution was degassed three times, and then Pd(PPh3)4(70 mg, 0.06 mmol) was added under the protection of nitrogen. The reaction was refluxed overnight. After removing the solvent, the crude product was purified by silica gel column chromatography(dichloromethane∶petroleum ether=1∶4) to obtain compound 4a as an orange oil (420 mg, 83%).
1H NMR(500 MHz, CDCl3),: 7.76(s, 2H), 7.05(s, 2H), 6.88(s, 2H), 4.31—4.19(m, 2H), 2.53 (d, 4H), 1.89—1.84(m, 1H), 1.63—1.59(m, 2H), 1.37—1.27(m, 40H), 0.93—0.87(m, 18H).13C NMR(125 MHz, CDCl3),: 161.84, 143.35, 136.34, 135.14, 134.68, 133.51, 128.28, 127.15, 122.06, 49.15, 38.86, 37.86, 34.92, 33.31, 33.00, 31.92, 30.83, 29.69, 28.88, 28.74, 26.61, 24.11, 23.15, 23.04, 22.71, 14.17, 14.15, 14.13, 10.74.
Compound 4b was prepared according to the similar procedures of compound 4a.
1H NMR(500 MHz, CDCl3),: 7.77(s, 2H), 7.05(s, 2H), 6.89(s, 2H), 3.60(s, 3H), 2.53(d, 4H), 1.64—1.59(m, 2H), 1.23—1.27(m, 32H), 0.91—0.87(m, 12H).13C NMR(125 MHz, CDCl3),: 161.62, 143.38, 136.43, 135.42, 134.60, 133.05, 128.08, 127.23, 122.13, 38.85, 34.92, 33.30, 33.06, 32.99, 31.92, 29.69, 28.87, 26.60, 23.05, 22.71, 14.17, 14.15.
Synthesis of 2,8-bis[5-bromo-4-(2-butyloctyl)thiophen-2-yl]-5-(2-ethylhexyl)-4H-dithieno[3,2-c∶2′,3′-e]azepine-4,6(5H)-dione(5a).
In a 50 mL two-necked flask, compound 4a(420 mg, 0.5 mmol) was dissolved in 30 mL chloroform, then-bromosuccinimide(178 mg, 1 mmol) was added and the reaction was stirred overnight at room temperature. After removing the solvent, the crude product was purified by silica gel column chromatography(dichloromethane∶petroleum ether=1∶2) to obtain compound 5a as an orange oil(410 mg, 82%).
1H NMR(500 MHz, CDCl3),: 7.71(s, 2H), 6.90(s, 2H), 4.31—4.18(m, 2H), 2.49(d, 4H), 1.90—1.79(m, 1H), 1.66(s, 2H), 1.36—1.22(m, 40H), 0.94—0.86(m,18H).13C NMR(125 MHz, CDCl3),: 160.25, 141.64, 134.14, 133.83, 133.32, 132.54, 127.46, 125.42, 109.86, 48.06, 37.50, 36.77, 33.15, 32.28, 32.00, 30.90, 29.80, 28.68, 27.74, 27.67, 25.48, 23.07, 22.14, 22.03, 21.70, 13.13, 13.11, 9.68.
Compound 5b was prepared according to the similar procedures of compound4a.
1H NMR(500 MHz, CDCl3, ppm),: 7.70(s, 2H), 6.89(s, 2H), 3.59(s, 3H), 2.48(d, 4H), 1.70—1.62(m, 2H), 1.32—1.25(m, 32H), 0.91—0.87(m, 12H).13C NMR(125 MHz, CDCl3),: 161.38, 142.77, 135.39, 135.34, 134.28, 133.21, 128.40, 126.67, 110.98, 38.54, 34.20, 33.30, 33.07, 33.02, 31.90, 29.67, 28.76, 26.50, 23.04, 22.70, 14.14.
Synthesis of polymer pBDT-BTI-EH. Compound 5a(100.6 mg, 0.10 mmol), BDT-DSn(90.5 mg, 0.10 mmol) and 2 mL chlorobenzene were added into a 15 mL Schlenk tube. Then Pd2(dba)3(2.7 mg, 0.003 mmol) and P(-Tol)3(9.1 mg, 0.03 mmol) were added into the tube under the protection of nitrogen. The reaction was refluxed for 36 h. After cooling to room temperature, the reaction solution was added dropwise to 150 mL of methanol. The precipitate was further extracted by Soxhlet extraction using acetone,-hexane, dichloromethane, and chloroform in sequence. The chloroform solution was concentrated and precipitated into methanol to give pBDT-BTI-EH as a purple-dark solid(78 mg, 55%). Thenfor pBDT-BTI-EH is 27100, with a PDI of 1.67.
Polymer pBDT-BTI-ME was prepared according to the similar procedures of pBDT-BTI-EH, in which the compound 5a was replaced by 5b. Thenfor pBDT-BTI-ME is 37900, with a PDI of 1.88.
2.3 Device Preparation of OSCs
The optimal OSCs with conventional device architecture of ITO/PEDOT∶PSS/active layer/PNDIT-F3N-Br/Ag were fabricated(Fig.1). First, PEDOT∶PSS was spin-coated(3500 r/min for 30 s) on the top of a cleaned ITO and annealed in air at 150 ℃ for 15 min to form a thin film of about 40 nm. Subsequently, optimized OSCs were prepared by using a mass ratio of polymer donor∶Y6=1∶1.2(polymer donor:6 mg/mL) with chloroform as processing solvent and 0.5%(volume fraction) 1-chloronaphthalene(CN) as solvent additive. Then the devices were thermally annealed at 100 ℃ for 10 min in a nitrogen protected glove-box. After that, a 10 nm PNDIT-F3N-Br layer was formed by spin-coating. Finally, a 100 nm Ag layer was deposited by thermal vacuum evaporation.
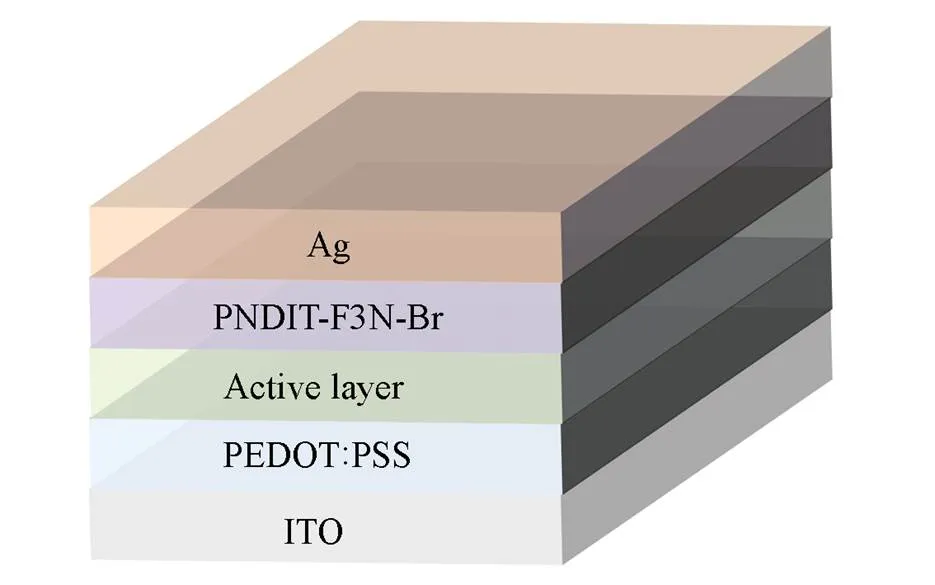
Fig.1 Device structure of OSCs
3 Results and Discussions
The molecular structures of polymer donors pBDT-BTI-EH and pBDT-BTI-ME with different lengths of side chains are shown in Fig.2(A) and (B). The polymer donor pBDT-BTI-EH demonstrates good solubility in chloroform(CF), chlorobenzene(CB), and-dichlorobenzene(-DCB) at room temperature. On the other hand, the polymer donor pBDT-BTI-ME with the shorter alkyl chain can be soluble in CB and-DCB. Thenand PDI of pBDT-BTI-EH were determined to be 27100 and 1.67, respectively, while pBDT-BTI-ME exhibited annof 37900 and a PDI of 1.88(Fig.S1, see the Supporting Information of this paper), as measured by high-temperature gel permeation chromatography(GPC). Furthermore, the 5% thermal weight decomposition temperatures(d) of pBDT-BTI-EH and pBDT-BTI-ME were found to be 406 ℃ and 432 ℃, respectively(Fig.S2, see the Supporting Information of this paper), indicating excellent thermal stability for both copolymers.
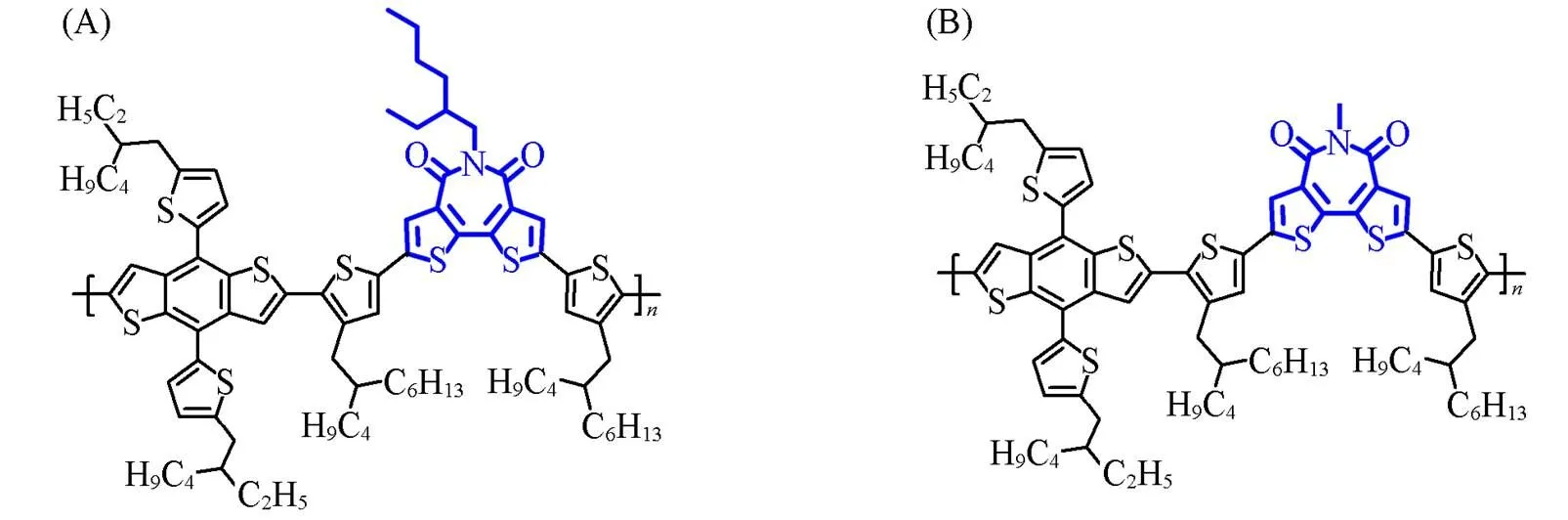
Fig.2 Chemical structures of pBDT⁃BTI⁃EH(A) and pBDT⁃BTI⁃ME(B)
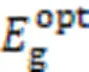
Fig.3 Normalized UV⁃Vis absorption spectra of pBDT⁃BTI⁃EH and pBDT⁃BTI⁃ME in chloroform solutions(A) and as thin films(B), temperature⁃dependent absorption spectra of pBDT⁃BTI⁃ME(C) and pBDT⁃BTI⁃EH(D) in chlorobenzene solution, cyclic voltammetry curves of polymer pBDT⁃BTI⁃EH and pBDT⁃BTI⁃ME(E) and energy level diagram of pBDT⁃BTI⁃EH, pBDT⁃BTI⁃ME and Y6(F)

Table 1 Optical properties and energy levels of pBDT-BTI-EH and pBDT-BTI-ME
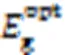
To further investigate the aggregation behaviors of two polymers, temperature-dependent UV-Vis absorption spectra of pBDT-BTI-ME and pBDT-BTI-EH in CB solutions were tested within a temperature range of 20—100 ℃. As depicted in Fig.3(C) and (D), the 0-0 peak observed in both pBDT-BTI-EH and pBDT-BTI-ME gradually diminished as the temperature increased, accompanied by a blue shift of the ICT peak. It indicates that the increase of temperature can disrupt the aggregation of the polymer chains. Notably, even when heated to 100 ℃, the 0-0 peak of pBDT-BTI-ME was not completely disappeared, while no 0-0 peak could be observed for pBDT-BTI-EH at about 80 ℃. These findings suggested that the polymer pBDT-BTI-ME with shorter side chains exhibits stronger aggregation behavior than pBDT-BTI-EH with longer side chains.
The highest occupied molecular orbital(HOMO) energy levels and the lowest unoccupied molecular orbital(LUMO) energy levels were estimated by cyclic voltammetry(CV) test. The CV curves are shown in Fig.3(E) and the corresponding HOMO/LUMO energy levels are summarized in Table 1. The HOMO/LUMO energy levels of ‒5.52/‒3.56 eV and ‒5.51/‒3.58 eV are determined for pBDT-BTI-EH and pBDT-BTI-ME, respectively. These energy levels are well matched with that of the non-fullerene acceptor Y6[Fig.4 and Fig.3(F)], affording the sufficient driving force for exciton dissociation.
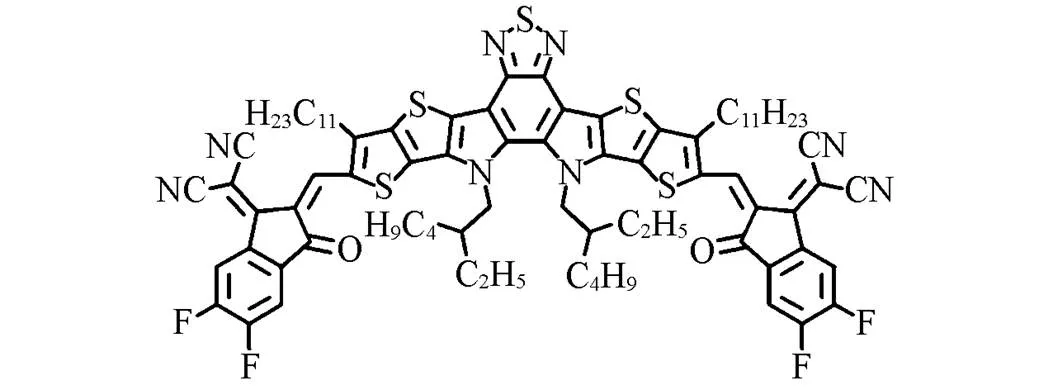
Fig.4 Chemical structure of Y6
OSCs were constructed based on a conventional device structure of ITO/PEDOT∶PSS/polymer donor∶Y6/PNDIT-F3N-Br/Ag(Fig.1) to investigate the photovoltaic performance of polymer donors with different alkyl side chains. Several factors, including solvent additives, spin-casting rates, donor: acceptor mass ratios, and annealing temperatures, were considered to optimize the photovoltaic performance of OSCs. Photovoltaic parameters of OSCs by optimization processes were shown in Table S1—Table S3(see the Supporting Information of this paper), while the optimized photovoltaic parameters of pBDT-BTI-EH/pBDT-BTI-ME based devices are listed in Table 2. Optimized OSCs were prepared by using a mass ratio of polymer donor∶Y6=1∶1.2(polymer donor: 6 mL) with CF as processing solvent and 0.5%(volume fraction) CN as a solvent additive. The devices were then thermally annealed at 100 ℃ for 10 min. The current density-voltage(-) characteristics of the optimized devices are shown in Fig.5(A). The optimized devices based on pBDT-BTI- EH∶Y6 exhibited a PCE of 9.31% with aOCof 0.87 V, aSCof 18.66 mA/cm2, and an FF of 56.43%. In contrast, due to the better charge transport and exciton dissociation properties, a much-enhancedSCof 25.48 mA/cm2and an FF of 73.20% were achieved for pBDT-BTI-ME based OSCs, leading to a remarkably higher PCE of 15.69%. Fig.5(B) illustrates the external quantum efficiency(EQE) spectra of pBDT-BTI-ME and pBDT-BTI-EH-based devices, both of which exhibited a broad photo-response from 300 nm to 900 nm. Furthermore, the EQE responses of pBDT-BTI-ME-based devices were much higher than that of pBDT-BTI-EH-based devices from 400 nm to 800 nm. The integratedSCvalues of pBDT-BTI-ME and pBDT-BTI-EH-based devices from the EQE spectra were 24.43 and 18.33 mA/cm2, respectively, which were consistent with those obtained from-characteristics within 5% deviation.
Table 2 Photovoltaic parameters of the optimized devices
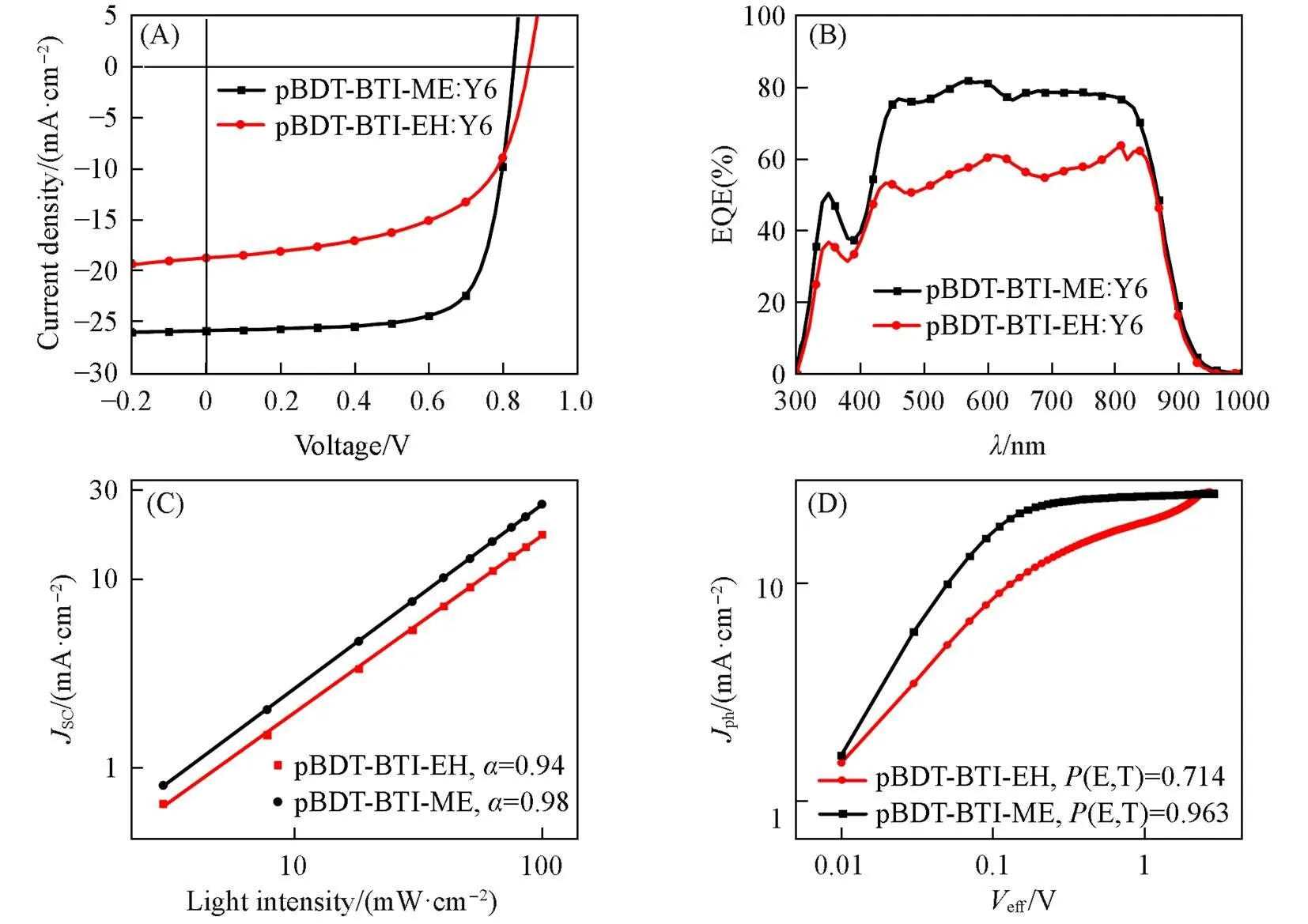
Fig.5 J⁃V curves(A), EQE spectra(B), the curves of JSCvs. light intensity(C) and Jphvs. Veff curves of pBDT⁃BTI⁃EH and pBDT⁃BTI⁃ME based devices(D)
To gain more insights into the enhanced PCE, exciton dissociation probabilities, charge recombination behaviors, and charge transport abilities were explored. TheSCandOCvaluesthe incident light intensity(light) were investigated to analyze the charge recombination mechanisms in devices. The relationship betweenSCandlightcan be correlated toSC∝light, where the exponential factor() indicates the degree of bimolecular recombination[59]. A value ofcloser to 1 indicates reduced bimolecular recombination. As shown in Fig.5(C), the α values of pBDT-BTI-EH and pBDT-BTI-ME-based devices were 0.94 and 0.98, respectively. This suggests that the pBDT-BTI-Me-based devices exhibited less bimolecular recombination, which was consistent with their higherSCand FF. To further understand the charge recombination mechanism in devices, the relationship betweenOCandlightwas studied, which could be described asOC∝klnlight[60]. A value ofcloser to 1 indicates reduced trap-assisted recombination. As shown in Fig.S3(see the Supporting Information of this paper), thevalues of pBDT-BTI-ME and pBDT-BTI-EH-based devices were 1.53 and 1.86, respectively. This indicates that the pBDT-BTI-ME-based devices exhibited less trap-assisted recombination.
The relationship between photocurrent density(ph) and effective voltage(eff) was investigated to further explore the exciton dissociation probabilities in devices. As shown in Fig.5(D), thephis defined asph=L‒D, whereLandDrepresent the current densities of devices under AM 1.5G illumination and dark conditions, respectively. Theeffis defined aseff=0‒bias, where0is the voltage whenph=0 mA/cm2andbiasis the applied bias voltage. The exciton dissociation probability(E,T) can be calculated from the equation(E,T)=ph,SC/sat, wheresatis the saturation photocurrent density at a high value ofeff>2 V andph,SCis the photocurrent density under short-circuit condition[61]. The calculated(E,T) of pBDT-BTI-EH and pBDT-BTI-ME-based devices were 71.4% and 96.3%, respectively. This indicates that the pBDT-BTI-ME based devices exhibited more efficient exciton dissociation, which is also consistent of the changing trend ofSCvalues. It is worth noting that thephof pBDT-BTI-EH-based devices does not saturate toat largeeffbut rather increases with largeeff, whereis the elementary charge,is the charge generation rates andis the thickness of the active layer. This fact was attributed to the electric field-dependent, which may be caused by the separation of geminate electron-hole pairs under a strong electric field[61—63].
To further understand the effect of different lengths of alkyl side chains on the charge transport capabilities, space-charge-limited current(SCLC) method was employed to obtain hole mobilities(h) and electron mobilities(e) of the blend films by fabricating electron-only and hole-only devices. The results were shown in Fig.S4(see the Supporting Information of this paper) and Table 2. The hole mobilities and electron mobilities of pBDT-BTI-ME-based device were 8.98×10‒3cm2·V‒1·s‒1and 2.35×10‒3cm2·V‒1·s‒1, respectively, which were much higher than those of pBDT-BTI-EH-based devices(h=1.33×10‒3cm2·V‒1·s‒1ande=2.11×10‒4cm2·V‒1·s‒1). The shorter alkyl chain in polymer donor pBDT-BTI-ME might induce stronger intermolecular interactions, resulting in a substantial increase in the charge mobilities. Furthermore, the pBDT-BTI-ME-based devices showed more balanced charge mobility(h/e=3.82) than that of pBDT-BTI-EH-based ones (h/e=6.30), which correlated with higher FF andSC.
The morphology of the active layer was investigated by AFM and transmission electron microscopy(TEM) to further explore the effects of the alkyl side chain on the microstructure within blend films. The AFM images of the pBDT-BTI-ME and pBDT-BTI-EH-based device are shown in Fig.6(A) and (B), and the root-mean-square roughness(q) of pBDT-BTI-ME∶Y6 and pBDT-BTI-EH∶Y6 blend films were determined to be 5.29 and 11.7 nm, respectively. In comparison, the pBDT-BTI-ME∶Y6 blend film exhibited a smoother surface. Fig.6(C) and(D) represent the TEM images of pBDT-BTI-ME∶Y6 and pBDT-BTI-EH∶Y6 blend films, respectively. In comparison, the pBDT-BTI-ME∶Y6 film exhibited a continuous and interpenetrated fibrous network with appropriate phase separation, which might be beneficial for charge transport and dissociation.

Fig.6 AFM images(A, B) and TEM images(C, D) of pBDT⁃BTI⁃ME∶Y6(A, C) and pBDT⁃BTI⁃EH∶Y6(B, D)
4 Conclusions
In this work, polymer donors pBDT-BTI-EH and pBDT-BTI-ME were constructed to investigate the effect of varying lengths of alkyl side chains on the photovoltaic properties. By altering the length of the alkyl side chains from 2-ethylhexyl to methyl, the intermolecular aggregation and packing could be strengthened. As fabricated into OSCs, the devices based on pBDT-BTI-ME∶Y6 exhibited higher and more balanced electron mobility, more effective exciton dissociation,lower charge recombination and more favorable film morphology, which collectively contributed to as significant increase in the PCE from 9.31% to 15.69%. This study underscores the importance of thoughtful design and precise tuning of alkyl side chains in polymer donors for high performance OSCs.
The supporting information of this paper see http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20230271.
[1] Lu L., Zheng T., Wu Q., Schneider A. M., Zhao D., Yu L.,,2015,(23), 12666—12731
[2] Zhang G., Zhao J., Chow P. C. Y., Jiang K., Zhang J., Zhu Z., Zhang J., Huang F., Yan H.,,2018,(7), 3447—3507
[3] Li Y.,,2012,(5), 723—733
[4] Zhang G., Lin F. R., Qi F., Heumüller T., Distler A., Egelhaaf H. J., Li N., Chow P. C. Y., Brabec C. J., Jen A. K. Y., Yip H. L.,,2022,(18), 14180—14274
[5] Yao H., Hou J.,,2022,(37), e202209021
[6] Huang F., Bo Z. S., Geng Y. H., Wang X. H., Wang L. X., Ma Y. G., Hu W. P., Pei J., Dong H. L., Wang S. Li Z., Shuai Z. G., Li Y. F, Cao Y.,,2019,(10), 988—1046(黄飞,薄志山,耿延候,王献红,王利祥,马於光,侯剑辉,胡文平,裴坚,董焕丽,王树,李振,帅志刚,李永舫,曹镛. 高分子学报,2019,(10), 988—1046)
[7] Liu C., Bai Y., Hu Z., Huang F.,,2022,(11), 1948—2000
[8] Cui Y., Xu Y., Yao H., Bi P., Hong L., Zhang J., Zu Y., Zhang T., Qin J., Ren J., Chen Z., He C., Hao X., Wei Z., Hou J.,,2021,(41), 2102420
[9] Chong K., Xu X., Meng H., Xue J., Yu L., Ma W., Peng Q.,,2022,(13), 2109516
[10] Zhu L., Zhang M., Xu J., Li C., Yan J., Zhou G., Zhong W., Hao T., Song J., Xue X., Zhou Z., Zeng R., Zhu H., Chen C. C., MacKenzie R. C. I., Zou Y., Nelson J., Zhang Y., Sun Y., Liu F.,,2022,(6), 656—663
[11] Pang B., Liao C., Xu X., Yu L., Li R., Peng Q.,,2023,(21), 2300631
[12] Fu J., Fong P. W. K., Liu H., Huang C. S., Lu X., Lu S., Abdelsamie M., Kodalle T., Sutter⁃Fella C. M., Yang Y., Li G.,,2023,(1), 1760
[13] Wang J., Wang Y., Bi P., Chen Z., Qiao J., Li J., Wang W., Zheng Z., Zhang S., Hao X., Hou J.,,2023,(25), 2301583
[14] Li S., Li C. Z., Shi M., Chen H.,,2020,(5), 1554—1567
[15] Yao H., Wang J., Xu Y., Zhang S., Hou J.,,2020,(4), 822—832
[16] Mishra A.,,2020,(12), 4738—4793
[17] Liu Y., Liu B., Ma C. Q., Huang F., Feng G., Chen H., Hou J., Yan L., Wei Q., Luo Q., Bao Q., Ma W., Liu W., Li W., Wan X., Hu X., Han Y., Li Y., Zhou Y., Zou Y., Chen Y., Li Y., Chen Y., Tang Z., Hu Z., Zhang Z. G., Bo Z.,,2022,(2), 224—268
[18] Song A., Huang Q., Zhang C., Tang H., Zhang K., Liu C., Huang F., Cao Y.,,2023,(8), 082202
[19] Park S.,Kim T., Yoon S., Koh C. W., Woo H. Y., Son H. J.,,2020,(51), 2002217
[20] Li Z., Ying L., Zhu P., Zhong W., Li N., Liu F., Huang F., Cao Y.,,2019,(1), 157—163
[21] Liu Y., Liu B., Ma C. Q., Huang F., Feng G., Chen H., Hou J., Yan L., Wei Q., Luo Q., Bao Q., Ma W., Liu W., Li W., Wan X., Hu X., Han Y., Li Y., Zhou Y., Zou Y., Chen Y., Liu Y., Meng L., Li Y., Chen Y., Tang Z., Hu Z., Zhang Z. G., Bo Z.,,2022,(8), 1457—1497
[22] Dong Y., Cha H.,Bristow H. L., Lee J., Kumar A., Tuladhar P. S., McCulloch I., Bakulin A. A., Durrant J. R.,,2021,(20), 7599—7603
[23] Karuthedath S., Gorenflot J., Firdaus Y., Chaturvedi N., De Castro C. S. P., Harrison G. T., Khan J. I., Markina A., Balawi A. H., Peña T. A. D., Liu W., Liang R. Z., Sharma A., Paleti S. H. K., Zhang W., Lin Y., Alarousu E., Anjum D. H., Beaujuge P. M., De Wolf S., McCulloch I., Anthopoulos T. D., Baran D., Andrienko D., Laquai F.,,2021,(3), 378—384
[24] Yuan J., Zhang Y., Zhou L., Zhang G., Yip H. L., Lau T. K., Lu X., Zhu C., Peng H., Johnson P. A., Leclerc M., Cao Y., Ulanski J., Li Y., Zou Y.,,2019,(4), 1140—1151
[25] Li S., Zhan L., Jin Y., Zhou G., Lau T. K., Qin R., Shi M., Li C. Z., Zhu H., Lu X., Zhang F., Chen H.,,2020,(24), 2001160
[26] Luo Z., Ma R., Chen Z., Xiao Y., Zhang G., Liu T., Sun R., Zhan Q., Zou Y., Zhong C., Chen Y., Sun H., Chai G., Chen K., Guo X., Min J., Lu X., Yang C., Yan H.,,2020,(44), 2002649
[27] Lai H., Zhao Q., Chen Z., Chen H., Chao P., Zhu Y., Lang Y., Zhen N., Mo D., Zhang Y., He F.,,2020,(3), 688—700
[28] Chen Y., Ma R., Liu T., Xiao Y., Kim H. K., Zhang J., Ma C., Sun H., Bai F., Guo X., Wong K. S., Lu X., Yan H.,,2021,(20), 2003777
[29] Chai G., Chang Y., Zhang J., Xu X., Yu L., Zou X., Li X., Chen Y., Luo S., Liu B., Bai F., Luo Z., Yu H., Liang J., Liu T., Wong K. S., Zhou H., Peng Q., Yan H.,,2021,(6), 3469—3479
[30] Chen Y., Bai F., Peng Z., Zhu L., Zhang J., Zou X., Qin Y., Kim H. K., Yuan J., Ma L. K., Zhang J., Yu H., Chow P. C. Y., Huang F., Zou Y., Ade H., Liu F., Yan H.,,2021,(3), 2003141
[31] Li C., Zhou J., Song J., Xu J., Zhang H., Zhang X., Guo J., Zhu L., Wei D., Han G., Min J., Zhang Y., Xie Z., Yi Y., Yan H., Gao F., Liu F., Sun Y.,,2021,(6), 605—613
[32] Chen S., Hong L., Dong M., Deng W., Shao L., Bai Y., Zhang K., Liu C., Wu H., Huang F.,,2023,(1), 202213869
[33] Zhu C., Yuan J., Cai F., Meng L., Zhang H., Chen H., Li J., Qiu B., Peng H., Chen S., Hu Y., Yang C., Gao F., Zou Y., Li Y.,,2020,(8), 2459—2466
[34] Zhou Z., Liu W., Zhou G., Zhang M., Qian D., Zhang J., Chen S., Xu S., Yang C., Gao F., Zhu H., Liu F., Zhu X.,,2020,(4), 1906324
[35] Liu S., Yuan J., Deng W. Y., Luo M., Xie Y., Liang Q. B., Zou Y. P., He Z. C., Wu H. B., Cao Y.,,2020,(5), 300—305
[36] Liang H., Chen H., Wang P., Zhu Y., Zhang Y., Feng W., Ma K., Lin Y., Ma Z., Long G., Li C., Kan B., Yao Z., Zhang H., Wan X., Chen Y.,,2023, doi: 10.1002/adfm.202301573
[37] Liu Q., Jiang Y., Jin K., Qin J., Xu J., Li W., Xiong J., Liu J., Xiao Z., Sun K., Yang S., Zhang X., Ding L.,,2020,(4), 272—275
[38] Cui C., Li Y.,,2019,(11), 3225—3246
[39] He K., Kumar P., Yuan Y., Li Y.,,2021,(1), 115—145
[40] Lee C., Lee S., Kim G. U., Lee W., Kim B. J.,,2019,(13), 8028—8086
[41] Hu Y., Li L., Wang X., Ma D., Huang F.,,2021,(3), 1017—1019
[42] Zhao W., Qian D., Zhang S., Li S., Inganäs, O., Gao, F., Hou, J.,,2016,(23), 4734—4739
[43] Fan B., Zhang K., Jiang X. F., Ying L., Huang F., Cao Y.,,2017,(21), 1606396
[44] Zhu C., Meng L., Zhang J., Qin S., Lai W., Qiu B., Yuan J., Wan Y., Huang W., Li Y.,,2021,(23), 2100474
[45] Wang Y., Yan Z., Guo H., Uddin M. A., Ling S., Zhou X., Su H., Dai J., Woo H. Y., Guo X.,,2017,(48), 15304—15308
[46] Shi Y., Guo H., Qin M., Zhao J., Wang Y., Wang H., Wang Y., Facchetti A., Lu X., Guo X.,,2018,(10), 1705745
[47] Feng K., Zhang X., Wu Z., Shi Y., Su M., Yang K., Wang Y., Sun H., Min J., Zhang Y., Cheng X., Woo H. Y., Guo X.,,2019,(39), 35924—35934
[48] Zhou N., Guo X., Ortiz R. P., Li S., Zhang S., Chang R. P. H., Facchetti A., Marks T. J.,,2012,(17), 2242—2248
[49] Zhou N., Guo X., Ortiz R. P., Harschneck T., Manley E. F., Lou S. J., Hartnett P. E., Yu X., Horwitz N. E., Burrezo P. M., Aldrich T. J., López Navarrete J. T., Wasielewski M. R., Chen L. X., Chang R. P. H., Facchetti A., Marks T. J.,,2015,(39), 12565—12579
[50] Sun H., Yu H., Shi Y., Yu J., Peng Z., Zhang X., Liu B., Wang J., Singh R., Lee J., Li Y., Wei Z., Liao Q., Kan Z., Ye L., Yan H., Gao F., Guo X.,,2020,(43), 2004183
[51] Li B., Yang K., Liao Q., Wang Y., Su M., Li Y., Shi Y., Feng X., Huang J., Sun H., Guo X.,,2021,(21), 2100332
[52] Chen W., Shi Y., Wang Y., Feng X., Djurišić A. B., Woo H. Y., Guo X., He Z.,,2020,, 104363
[53] Feng K., Guo H., Wang J., Shi Y., Wu Z., Su M., Zhang X., Son J. H., Woo H. Y., Guo X.,,2021,(3), 1539—1552
[54] Xu J., Feng H., Liang Y., Tang H., Tang Y., Du Z., Hu Z., Huang F., Cao Y.,,2022,, 382—389
[55] Letizia J. A., Salata M. R., Tribout C. M., Facchetti A., Ratner M. A., Marks T. J.,,2008,(30), 9679—9694
[56] Guo X., Zhou N., Lou S. J., Hennek J. W., Ponce Ortiz R., Butler M. R., Boudreault P. L. T.,Strzalka J., Morin P. O., Leclerc M., López Navarrete J. T., Ratner M. A., Chen L. X., Chang R. P. H., Facchetti A., Marks T. J.,,2012,(44), 18427—18439
[57] Wang G., Swick S. M., Matta M., Mukherjee S., Strzalka J. W., Logsdon J. L., Fabiano S., Huang W., Aldrich T. J., Yang T., Timalsina A., Powers⁃Riggs N., Alzola J. M., Young R. M., DeLongchamp D. M., Wasielewski M. R., Kohlstedt K. L., Schatz G. C., Melkonyan F. S., Facchetti A., Marks T. J.,,2019,(34), 13410—13420
[58] Bai Y., Zhou Z., Xue Q., Liu C., Li N., Tang H., Zhang J., Xia X., Zhang J., Lu X., Brabec C. J., Huang F.,,2022,(49), 2110587
[59] Schilinsky P., Waldauf C., Brabec C. J.,,2002,(20),3885—3887
[60] Koster L. J. A., Mihailetchi V. D., Ramaker R., Blom P. W. M.,,2005,(12), 123509
[61] Mihailetchi V. D., Koster L. J. A., Hummelen J. C., Blom P. W. M.,,2004,(21), 216601
[62] Proctor C. M., Kim C., Neher D., Nguyen T. Q.,,2013,(28), 3584—3594
[63] Jia T., Zhang J., Zhang K., Tang H., Dong S., Tan C. H., Wang X., Huang F.,,2021,(14), 8975—8983
基于双噻吩酰亚胺聚合物给体的侧链调控与光伏性能研究
白原青#,张佳滨#,刘春晨,胡志诚,张凯,黄飞
(华南理工大学发光材料与器件国家重点实验室, 高分子光电材料与器件研究所, 广州 510640)
采用苯并二噻吩(BDT)作为给电子单元, 分别与具有2-乙基己基和甲基侧链的双噻吩酰亚胺(BTI)缺电子单元共聚构筑了两个聚合物给体材料(pBDT-BTI-EH和pBDT-BTI-ME). 与pBDT-BTI-EH∶Y6相比, 基于 pBDT-BTI-ME∶Y6的器件具有更高的电荷迁移率、更低的载流子复合、更高的激子解离以及更优的薄膜形貌, 从而获得了更高的短路电流密度(SC)和填充因子(FF), 电池的能量转换效率由9.31%提高到15.69%.
有机太阳电池;聚合物给体;双噻吩酰亚胺;侧链调控
O631
A
10.7503/cjcu20230271
2023-06-06
网络首发日期: 2023-07-79.
联系人简介:黄飞, 男, 博士, 教授, 主要从事有机光电材料及器件研究. E-mail: msfhuang@scut.edu.cn
刘春晨, 男, 博士, 副研究员, 主要从事有机光电材料及器件研究. E-mail: mscliu@scut.edu.cn
国家重点研发计划项目(批准号: 2019YFA0705900)、广东省基础与应用基础研究重点项目(批准号: 2019B030302007)、国家自然科学基金(批准号: U21A6002)和粤港澳光电磁功能材料联合实验室(批准号: 2019B121205002)资助.
Supported by the National Key Research and Development Program of China(No.2019YFA0705900), the Basic and Applied Basic Research Major Program of Guangdong Province, China(No.2019B030302007), the National Natural Science Foundation of China(No.U21A6002) and the Guangdong-Hong Kong-Macao Joint Laboratory of Optoelectronic and Magnetic Functional Materials, China(No.2019B121205002).
# 共同第一作者.
(Ed.: V, K, S)
猜你喜欢
山东农业大学学报(自然科学版)(2021年3期)2021-07-29 03:07:02
民用飞机设计与研究(2020年1期)2020-05-21 07:24:52
世界农药(2019年2期)2019-07-13 05:55:12
中国塑料(2017年2期)2017-05-17 06:13:21
粘接(2017年4期)2017-04-25 08:37:20
当代化工研究(2016年1期)2016-03-16 22:00:24
合成化学(2015年10期)2016-01-17 08:56:47
原子与分子物理学报(2015年3期)2015-11-24 12:49:34
中国塑料(2015年6期)2015-11-13 03:02:49
应用化工(2014年9期)2014-08-10 14:05:08

- 高等学校化学学报的其它文章
- 高性能半透明有机太阳能电池的实现途径
- Beta-alanine as a Dual Modification Additive in Organic Solar Cells
- 半透明有机太阳能电池研究进展
- Aggregation Morphology of Perylene Bisimide Acceptors and the Role on Exciton Processes and Electron Transport in Organic Solar Cells
- 协同富勒烯和非富勒烯受体提高卟啉全小分子三元有机太阳能电池的性能
- 基于受体1-受体2型聚合物给体的高效有机太阳能电池