
湘潭地区异常血红蛋白病的分子特征与表型分析*
2023-10-08 09:35:14王淑媛殷伟黄京希李晨辉周玉珍刘益清
广东医学 2023年9期
王淑媛, 殷伟, 黄京希, 李晨辉, 周玉珍, 刘益清
湘潭市妇幼保健院优生遗传科(湖南湘潭 411104)
血红蛋白病是一类珠蛋白基因变异而导致血红蛋白结构和功能异常的遗传病[1],包括异常血红蛋白病和地中海贫血两大类,以热带和亚热带地区分布较广。以往针对湖南地区人群的地中海贫血相关报道较多,但对异常血红蛋白病、尤其是罕见的异常血红蛋白病的发生情况报道较少。本研究通过对2016年7月至2020年6月收集的湖南湘潭地区76例异常血红蛋白病进行分析,旨在分析本地区异常血红蛋白病的分布特征,探讨异常血红蛋白病的分子生物学特征和临床表型、为该地区人群血红蛋白病的筛查和遗传咨询提供理论依据。
1 资料与方法
1.1 一般资料 收集2016年7月至2020年6月在湘潭市妇幼保健院进行婚前检查、孕期检查的正常人群及非血液病门诊、住院患者作为研究对象,共收集连续性资料样本25 516例,年龄0~45岁,其中女24 677例,男839例。纳入标准:(1)在湘潭市妇幼保健院进行婚前检查、孕期检查的正常人群及非血液病门诊、住院患者。(2)同意采集静脉血2~4 mL,用于进行血细胞参数检测、血红蛋白电泳分析、地贫基因检测和珠蛋白基因序列分析。(3)静脉血标本经EDTA抗凝,于2~8℃中妥善保存。排除标准:(1)合并除地贫外的血液学疾病;(2)合并内科慢性贫血性疾病;(3)恶性肿瘤。参与研究对象均签署知情同意书,本研究已经过湘潭市妇幼保健院伦理委员会批准(202011)。
1.2 血液学表型分析 采用BC-6800全自动血液细胞分析仪(深圳迈瑞)进行血细胞参数检测。采集并分析血常规参数:血红蛋白含量(Hb)、平均红细胞体积(MCV)、平均红细胞血红蛋白含量(MCH)。采用Capillarys 2(Sebia,法国)全自动毛细管电泳仪及配套试剂进行血红蛋白电泳分析,分区(Z1-Z15区)鉴别血红蛋白条带类型,并分析各条带所占相对比例含量。针对血液学检测结果,符合以下条件之一的行常见α、β地贫基因检测:(1)血细胞参数检测MCV<80 fL;(2)MCH<27 pg;(3)血红蛋白电泳分析Hb A2≤2.5%或Hb A2≥3.5%;符合以下条件之一的行珠蛋白基因Sanger测序:(1)血红蛋白电泳出现异常条带;(2)常见α、β地贫基因阴性但血液学参数异常。
1.3 常见α、β地中海贫血基因检测 采用PCR结合反向斑点杂交技术(RDB)检测α地贫的3种常见缺失类型(--SEA、-α3.7和-α4.2)和3种常见点突变(αCSα、αQSα和αWSα)、β地贫的17种常见点突变(41-42M、654M、-28M、71-72M、17M、βEM、IVS-I-1M、IVS-I-5M、27/28M、43M、-29M、-30M、31M、-32M、14-15M、IntM和CAPM)。所用试剂为亚能生物技术(深圳)有限公司提供的地中海贫血基因检测试剂盒(注册证编号:国械注准20153401272)。操作程序按照试剂盒说明书进行。
1.4 珠蛋白基因测序 采用PCR方法扩增α1珠蛋白基因、α2珠蛋白基因和β珠蛋白基因, 采用Sanger双脱氧链终止法进行珠蛋白基因序列测定,于人类异常血红蛋白和地中海贫血数据库(http://globin.cse.psu.edu/)查找突变位点。珠蛋白基因测序均由亚能生物技术(深圳)有限公司检测。
2 结果
2.1 珠蛋白基因测序结果 在25 516例样本中,检出异常血红蛋白病76例,血红蛋白异常检出率0.29%(76/25 516),其中位于α珠蛋白肽链变异类型6种共30例,占比39.5%(30/76),最常见的α珠蛋白变异类型为Hb CS,共24例,占31.6%(24/76),其次为Hb Queens,共2例占比2.63%(2/76),1例少见型异常血红蛋白Hb Port Phillip(图1-A)及1例Hb Woodville合并Hb G-Coushatta异常血红蛋白(图2-A、B),另外还检测到1例Hb Q-Thailand 复合-α4.2杂合子;位于β珠蛋白肽链的变异类型6种共46例,占比59.2%(46/76),最常见的β珠蛋白变异类型为HbE,共16例,占21.1%(16/76),其次为Hb New York,共12例,占15.79%(12/76);α合并β珠蛋白肽链变异类型1例,占比1.3%(1/76)。异常血红蛋白变异类型见表1。

表1 异常血红蛋白变异类型及例数、构成比

注:A:珠蛋白基因测序(HBA2:c.275T>C);B:血红蛋白毛细管电泳图

注:A:珠蛋白基因测序(HBA1:c.19G>T);B:珠蛋白基因测序(HBB:c.68A>C);C:血红蛋白毛细管电泳图
2.2 常见α、β地中海贫血基因检测结果 在76例异常血红蛋白样本中,经PCR结合反向斑点杂交技术检出异常血红蛋白病40例,检出率52.6%(40/76)。包括Hb CS 24例,Hb E 16例,其中1例Hb CS复合β地贫杂合子,1例Hb E复合-α3.7杂合子。由于Hb CS、Hb E既会导致类α与类β珠蛋白比例失衡出现地中海贫血,又会导致异常血红蛋白产生,本研究中将Hb CS、Hb E纳入异常血红蛋白病讨论。
2.3 毛细管电泳结果 76例异常血红蛋白病的电泳结果发现异常血红蛋白条带(Hb X)70例,检出率92.11%(70/76)。30例α链异常血红蛋白病中,6例Hb CS没有检出异常条带(5例单纯Hb CS杂合子和1例Hb CS复合β地贫),24例检出异常条带。一例少见型异常血红蛋白Hb Port Phillip电泳图,在Z8区有3.7%异常条带(图1-B)。Hb CS电泳带含量普遍较低,在0.5%~1.2%。45例β链异常血红蛋白病均检出异常条带(表2)。一例Hb Woodville合并Hb G-Coushatta异常血红蛋白电泳图,在Hb S及Hb D区分别有3%和38.1%异常条带(图2-C)。76例异常血红蛋白病中42例Hb A2值偏离参考区间。α链异常血红蛋白病表现为Hb A2含量降低(<2.5%),有26例,占比86.7%(26/30);β链异常血红蛋白病表现为Hb A2升高(>3.5%),有16例,占比35.6%(16/45),全部为Hb E(表2)。

表2 异常血红蛋白病毛细管电泳检测情况 例(%)
2.4 血常规参数 在13种异常血红蛋白类型中,有3种(Hb CS、Hb E和Hb Port Phillip)表现为血常规参数异常,其他9种类型异常血红蛋白的单纯杂合子血常规参数均正常(表3)。单纯Hb CS杂合子平均Hb、MCV、MCH在正常范围,其中有6例Hb<110 g/L,5例MCH异常,1例MCV异常。1例Hb CS杂合子复合β地贫,其血常规为中度小细胞低色素性贫血。单纯Hb E杂合子表现为轻度β地中海贫血的血液学特征,Hb E杂合子复合-α3.7杂合子,其Hb、MCV和MCH水平较单纯Hb E杂合子异常程度减轻。Hb Q-Thailand合并-α4.2杂合子表现为类似静止型α地贫的特征;Hb Port Phillip杂合子表现为类似轻型α地贫的特征。
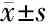
表3 异常血红蛋白的分子特征及血液学表型
3 讨论
异常血红蛋白病是由于珠蛋白基因缺陷导致珠蛋白分子的一级结构发生改变引起的一组遗传性血红蛋白病[2]。基于二代测序技术平台的湖南省血红蛋白病的流行病学调查研究显示,湖南省14个地市异常血红蛋白病平均发生率为0.49%[3],本研究通过联合血常规、血红蛋白电泳、常见地贫基因检测、Sanger测序4种技术的方式发现湘潭地区异常血红蛋白病携带率为0.298%,提示湖南地区异常血红蛋白病分布存在地区差异性。本研究检测结果显示β链异常血红蛋白病检出数多于α链异常血红蛋白病。以Hb CS和Hb E最为常见,其次为Hb New York、Hb G-Coushatta和Hb J-Bangkok。其中Hb CS、Hb E、Hb NewYork和Hb J-Bangkok是南方地区最常见异常血红蛋白病[4-7],Hb G-Coushatta在北方常见[8]。湘潭地区异常血红蛋白基因有其地区特异性,既有较高比例的南方人群常见的异常血红蛋白类型,也有北方人群常见的类型。
本研究中的76例异常血红蛋白病中,毛细管电泳检出异常血红蛋白条带70例,检出率92.11%;其中24例Hb CS中有18例毛细管电泳检出异常Hb带,检出率75%,未检出异常血红蛋白条带的6例,可能是由于Hb CS化学性质不稳定,在电泳上机之前发生了降解[9]。本研究中α链异常血红蛋白中有86.7%的个体Hb A2值偏离参考区间(<2.5%),而β链异常血红蛋白中仅有35.6%的个体Hb A2值异常(>3.5%)。因为Hb A2由α和δ珠蛋白组成,α链异常导致正常α珠蛋白数量减少会直接影响Hb A2的量,而β链异常是通过使α珠蛋白过剩或者δ珠蛋白代偿性表达上升间接影响Hb A2的量,当β链异常导致上述两种情况才会表现Hb A2偏高[10],当出现Hb E时,由于β珠蛋白基因突变导致异常血红蛋白Hb E合成的同时致使正常β珠蛋白表达减少而导致α珠蛋白过剩[11],本研究中Hb E异常血红蛋白病也表现出了Hb A2偏高的情况。同时缺铁性贫血会导致Hb A2表达水平降低而掩盖部分β链异常导致的Hb A2异常[12],因此,α链异常血红蛋白病表现出Hb A2指标异常概率较大,β链异常血红蛋白病表现出Hb A2指标异常概率较小,值得临床关注。
从血常规结果观察,大多数的单纯异常血红蛋白病血常规无异常表型,Hb CS、Hb E、Hb Port Phillip杂合子可表现为Hb、MCV、MCH参数异常,类似轻型地中海贫血的表现。本研究中检测结果显示,23例单纯Hb CS杂合子的血液学表型正常或接近正常。官志扬等[13]报道,Hb CS杂合子对β地中海贫血的血液学参数无显著影响,轻型β地贫的表现可掩盖Hb CS杂合子的存在。本研究中1例Hb CS杂合子复合β地贫,表现为β地贫的血液学特点,但贫血程度加重,为中度贫血。分析可能是由于个体差异或该病例为孕妇存在生理性贫血的可能。提示当Hb CS合并地中海贫血时表现可能有较大差异,同时Hb CS稳定差,在杂合子中含量低,所以Hb CS杂合子通过血常规和血红蛋白电泳筛查可能出现漏筛,在临床咨询中应加以重视。本研究中检出Hb E 16例,15例Hb E杂合子的血液学表型与β+地中海贫血相似,1例Hb E合并α+地贫,其血液学表型与单纯Hb E杂合子相似但更轻微。这是因为β珠蛋白基因26位密码子突变导致Hb E合成的同时会致使β珠蛋白表达减少,当Hb E合并α+地贫时,α和β珠蛋白表达同时减少,一定程度上缓解了单纯Hb E或α+地贫时α和β珠蛋白的不平衡[11]。该病例提示Hb E的出现会掩盖α+地贫的存在,临床分析时应引起注意。Hb Port Phillip是α链异常血红蛋白病,首次报道见于1977年新西兰1例华裔女性[14],国内深圳[15]曾报道一例出生10日龄新生儿,血液分析提示小细胞低色素性贫血,未行血红蛋白电泳检测。本研究中Hb Port Phillip病例为一个8个月的男孩,Hb电泳在Z8区有3.7%异常条带,Hb A2在正常范围,轻度小细胞低色素性贫血,Hb Port Phillip是一种可以引起α地中海贫血表型的异常血红蛋白[15],当其复合地贫尤其是α地贫时可能加重原有地贫的临床表型。因此,在临床遗传咨询时应加以注意。
其他少见型异常血红蛋白中,Hb Woodville首次于越南报道[16],为1例无异常临床表现及血液学表型的妇女,发现有9%异常血红蛋白。国内目前仅有佛山报道[17]1例。本例为Hb Woodville杂合子复合Hb G-Coushatta杂合子突变,无异常临床表现,电泳发现3%和38.1%异常条带,单纯Hb Woodville杂合突变的异常带含量一般为9%[16],而Hb Woodville血红蛋白既可能由α1基因突变导致,又可能由α2基因突变导致,且α2基因是α1基因表达量的2倍[11],本例因为突变基因位于α1基因使得异常条带含量较低。单纯Hb Woodville或Hb G-Coushatta杂合突变氨基酸残基位于血红蛋白的外部,未影响到血红蛋白结构及功能,因此,血液学表型无血常规异常,在毛细管电泳显示有异常血红蛋白条带。此外,本研究检出1例Hb Q-Thailand复合-α4.2地贫基因,除有28.4%异常Hb带外,其他血液学表型与-α4.2地贫无明显差异,与既往文献[18-19]报道一致。
多数杂合子异常血红蛋白病并没有明显的临床症状,少数异常血红蛋白可以引起地中海贫血的表型,尤其当这些异常血红蛋白复合地贫时可改变原有的临床表型。因此在血红蛋白电泳筛查出异常条带后,有必要对异常血红蛋白病进行基因检测确诊,特别是对孕妇或计划生育的夫妇根据不同的异常血红蛋白病基因型,进行遗传咨询和生育指导。
利益相关声明:不存在竞争经济利益。
作者贡献说明:黄京希、李晨辉、周玉珍、刘益清负责资料收集和整理;王淑媛、殷伟负责方案设计、数据统计与分析、论文撰写。
猜你喜欢
中国毕业后医学教育(2022年1期)2022-08-19 02:51:34
检验医学与临床(2021年14期)2021-07-29 07:40:24
莫愁(2019年34期)2020-01-01 02:18:10
伴侣(2019年7期)2019-07-25 06:34:47
莫愁·智慧女性(2019年12期)2019-06-01 10:12:59
湖北农业科学(2015年11期)2015-07-31 08:21:47
检验医学与临床(2015年14期)2015-03-16 01:46:53
现代检验医学杂志(2015年1期)2015-02-06 01:59:18
实验动物与比较医学(2014年3期)2014-02-28 14:52:55
卫生职业教育(2014年8期)2014-02-16 08:00:16
