
特殊浸润性材料在防治油气管道中水合物成核与聚集的应用
2023-10-07 12:34:48尹新宇皮丕辉文秀芳钱宇
化工进展 2023年8期
尹新宇,皮丕辉,文秀芳,钱宇
(广东省绿色化学品重点实验室,华南理工大学化学与化工学院,广东 广州 510640)
天然气水合物广泛存在于海床和多年冻土地区,是极具潜力的清洁能源[1]。然而,随着人类对油气资源的需求日益增加,油气开采已经转向油气资源更加丰富的深海。在深海油气开采运输过程中,高压低温条件为水合物的形成提供了有利的条件,水合物颗粒随着流体流动并逐渐形成体积更大的水合物团簇,水合物团簇的长期聚集沉积不仅对生产设备以及人身安全造成严重威胁,严重时将导致巨大经济损失和环境污染,这对油气开采、储运带来了极大挑战。因此,如何保障油气资源安全开采和储运成为亟待解决的难题。目前防治水合物堵塞的传统方法分为物理方法和化学方法。物理方法,例如管道伴热、机械刮擦等,存在能耗大、效率低、有安全隐患等问题。化学方法主要是通过注入化学抑制剂抑制或减缓水合物生长聚集,简单高效,但是存在生产成本高,分离困难,危害环境以及环境适应性差等问题。近年来特殊润湿性材料在自清洁、防黏附、防结冰、减阻等领域都展现巨大应用潜力,然而其在油气管道中的应用被长期低估。本文综述了水合物成核聚集堵塞过程以及影响因素,结合特殊润湿性材料的特点,并对特殊润湿性材料在抑制水合物以及防水合物黏附相关研究进行汇总,最后对特殊润湿性材料在抑制水合物成核、防水合物黏附中的应用进行展望。
1 水合物成核、聚集过程以及影响因素
1.1 水合物基本性质
天然气水合物广泛存在于海床和多年冻土地区,1m³水合物可以存储160~180m³天然气和0.8m³左右的水,是一种极具潜力、尚未大规模开发的绿色清洁能源。天然气水合物的形成通常是在低温高压条件下,小分子(客体分子),例如甲烷、乙烷等小分子烃类,被封闭在由水分子(宿主分子)通过氢键构建的多种笼形结构中[2-3],如图1(a)所示。
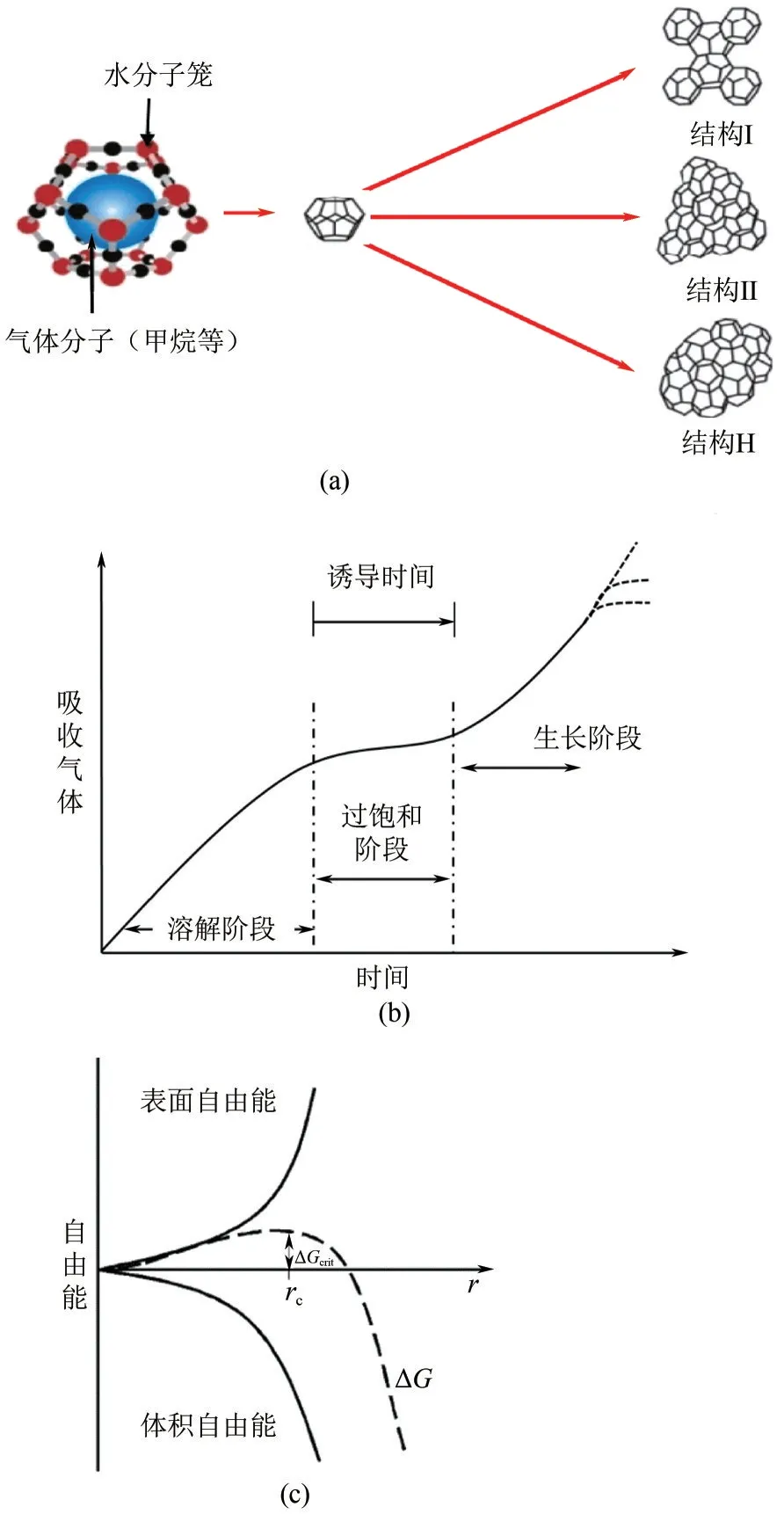
图1 常见水合物结构(a)、成核过程(b)和成核自由能(c)[4]
天然气水合物形成主要受温度、压力、相组成和相界面接触面积等因素影响。图1(b)表示了水合物形成过程中的三个主要阶段:溶解阶段、过饱和阶段和生长阶段。水合物成核的驱动力被认为是过冷度,也有认为成核的驱动力是水合物相中水的化学势和水相中水的化学势的差异,如图1(c)所示。水合物随机成核行为受到驱动力强度的强烈影响,这使得水合物成核有两种类型:均匀成核和异相成核[5-7]。均匀成核:通常在没有任何杂质的系统中观察水合物均匀成核,本质上是随机的,即由于系统的局部热力学波动而形成临界核。异相成核:主、客体分子与第三相接触成核,第三相可以是外来颗粒或基材表面。相比均相成核,异相成核的临界半径更小,更容易成核。
1.2 水合物成核、聚集堵塞过程
石油主导和天然气主导系统中水合物形成过程[4,8],如图2所示。在天然气水合物成核生长过程中,质量和热量传递是主要因素。对于水中溶解度较低的气体,例如,大多数天然气组分,质量传递更为重要。客体分子在大量水中的溶解度通常是每千个水分子中的几个分子,其太小而不能提供足以引发水合物形成的热力学驱动力。因此,水合物均相成核通常是困难的。但界面处通常具有非常高的主体和客体分子浓度,因而天然气水合物的形成通常在气-水或气-固界面处开始。
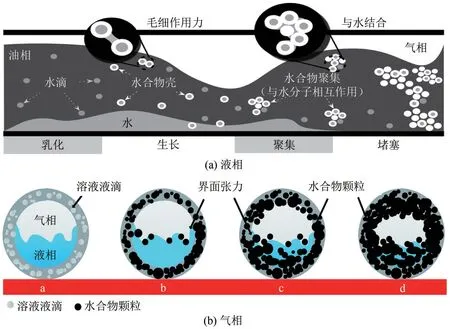
图2 液相和气相主导体系中水合物成核、聚集过程[4,8]
1.2.1 水合物成核过程以及成核位点
对不同体系成核位置不同,成核过程也不同。可溶性客体分子成核过程,如图3(a)所示。Florusse等[9]研究表明可溶性客体分子成核首先是形成水合物状薄膜,然后溶解的气体分子进入膜内,最终形成天然气水合物。此外,研究表明在埃米级粗糙表面水分子与甲烷气体分子接触就会触发水合物成核,即成核可发生在界面处且水合物优先在界面处成核[10-11]。对于不可溶性客体分子成核过程,如图3(b)所示,首先在烃-水界面水合物形成多孔薄膜,薄膜沿相界面成核。Taylor等[12]检测到环戊烷(CyC5)和甲烷水合物在烃-水界面水合物膜的生长以及水合物在水相中成核过程。Rodger等[13]通过分子动力学(MD)模拟表明成核位点是甲烷和二氧化碳水合物的气-液界面,进一步证实天然气水合物成核发生在烃-水界面处[14]。
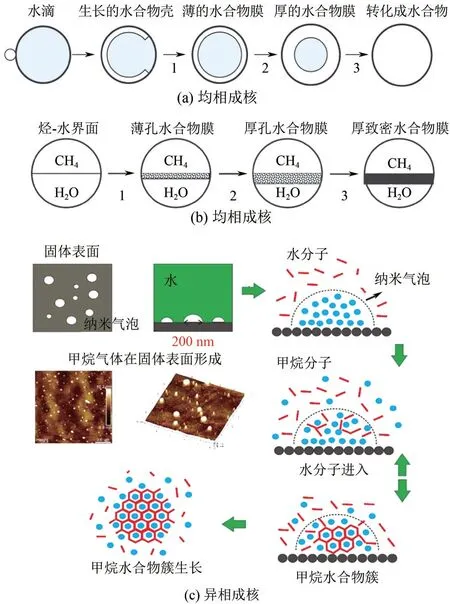
图3 不同体系中水合物成核过程[15]
以上两种成核方式均可以看做是均相成核,实际上异质成核才是水合物成核的主要途径。Bai等[16]研究发现水合物的成核发生在三相接触线附近,并沿三相接触线生长。Guo 等[15]提出甲烷首先在疏水性固-水界面上形成了气泡,为甲烷水合物的形成提供了成核位点。如图3(c)所示,甲烷纳米气泡在固体表面形成,甲烷分子与水分子相互作用形成笼形结构,逐渐成核,核大小超过临界尺寸后快速生长。此外,Zhang 等[17]研究表明冰可以吸收水合物形成所释放的热量,冰面的吸附作用促使其周围甲烷浓度较高,冰面与水合物笼之间的氢键相互作用导致水合物异相成核优先在冰表面附近而非本体液相中发生,而冰的亲水性和冰晶格与笼形水合物结构不匹配,可以抑制冰面上水合物异质成核,并且促进均匀成核。Nguyen等[18]研究表明在相同的热力学条件下,界面过渡层中的水具有比冰或包合物低且比液态水高的四面体有序度,界面过渡层可以提供比冰-液和水合物-液体界面更低的表面能来促进水合物异质成核。Odendahl等[19]首次发现水-空气界面竟然存在类冰有序结构,进一步佐证了界面处水分子有序排列促进水合物成核。
相比于二维固体表面,具有高表面积和更高传质传热性能的三维微纳米颗粒更容易促进水合物成核[20-21]。这是因为微纳米颗粒在流体中的布朗运动可以充当搅拌器并减少了气/水界面的薄膜阻力,强化传质,增强成核驱动力[22]。气体水合物形成是放热过程,放热产生的热量导致体系温度升高,降低了水合物形成的驱动力。通过添加导热性能更强的固体颗粒可以增加液体的导热率,可以更高效地从系统中除去成核释放的热量[23]。例如,氧化铜和氧化银等金属颗粒以及非金属MWCNT 等[24-28]在增加成核表面积的同时还能够增强体系的传热作为异相成核的种子,降低成核诱导时间。与纳米颗粒类似,微纳米多孔介质同样具有较大的表面积,能够增加体系的传质面积和传热潜力[29-32]。例如,碳化硅(SiC)可以将诱导时间缩短88%,成核速率降低95%,同时水合物转化率提高1.2 倍[33]。类似的蜂窝状金属铝,与添加十二烷基硫酸钠(SDS)体系相比,最大气体消耗率增加了9.62%~14.30%[34]。开孔泡沫铜的有效导热系数导致即便是在低压条件下仍然可以表现出更高的甲烷储存容量和更大的水合速率[7]。活性炭腔内的限制作用还可以改变局部甲烷的浓度并促进水合物的形成[35]。
显然,第三相的存在提供更多的成核位点[15,24,36],强化传热传质[37-38],加速水合物成核过程[39]。这也为抑制水合物成核提供思路,通常抑制浓度越高抑制性能越强,相比增大抑制剂浓度或增加改性抑制纳米粒子用量,若将抑制剂固定在具有高比表面积的三维多孔材料表面既可以抑制增强抑制性能,同时解决了使用化学抑制剂或者改性纳米粒子等方法带来的分离回收、污染环境等问题。
1.2.2 水合物聚集沉积过程及影响因素
在油相、水相、气相主导体系下水合物晶核生长聚集过程,如图4(a)所示。在油相中,水合物首先在油-水界面形成水合物壳,随后受泵流速和含量水的影响,壳生长和水合物输送能力的降低,从而导致水合物堵塞管道[3];在水相中,水合物均相成核,水合物浓度增加导致水合物床层和壁面沉积物增多,最终导致管道堵塞[43];在气相中,水合物将沉积在管壁上,从成核到树枝状生长,最终沉积物导致管道堵塞[44]。Ding 等[40]研究了不同流动模式中的水合物流动形态和浆料堵塞机理,如图4(b)所示,结果表明从高到底团聚程度:团状流>分层流>泡沫流>环状流;沉积程度从高到低依次为环形流动>团状流动>气泡流动>流体流动。通过观察乳液体系中的水合物沉积过程研究发现,水合物沉积过程受水合物驱动力、管道表面性质、含水量、传质系数和流动剪切力的影响。Aman 等[42]研究表明涂层可用于控制管道表面的水合物凝聚和沉积。在水相中,固-固黏附是主要黏附机制,而固体-固体黏附力、水合物-基材黏附机制由分离产生的界面面积和固-液界面张力决定;在油相中,通过液桥结合水合物颗粒所产生的毛细管黏附是主要机制,而毛细管黏附力取决于固-水界面张力、固-油界面张力、水-油界面张力,是三个界面张力的函数;水合物与基材之间的黏附力与水合物颗粒间的黏附机制相同,均随表面能的增加而增强[45]。
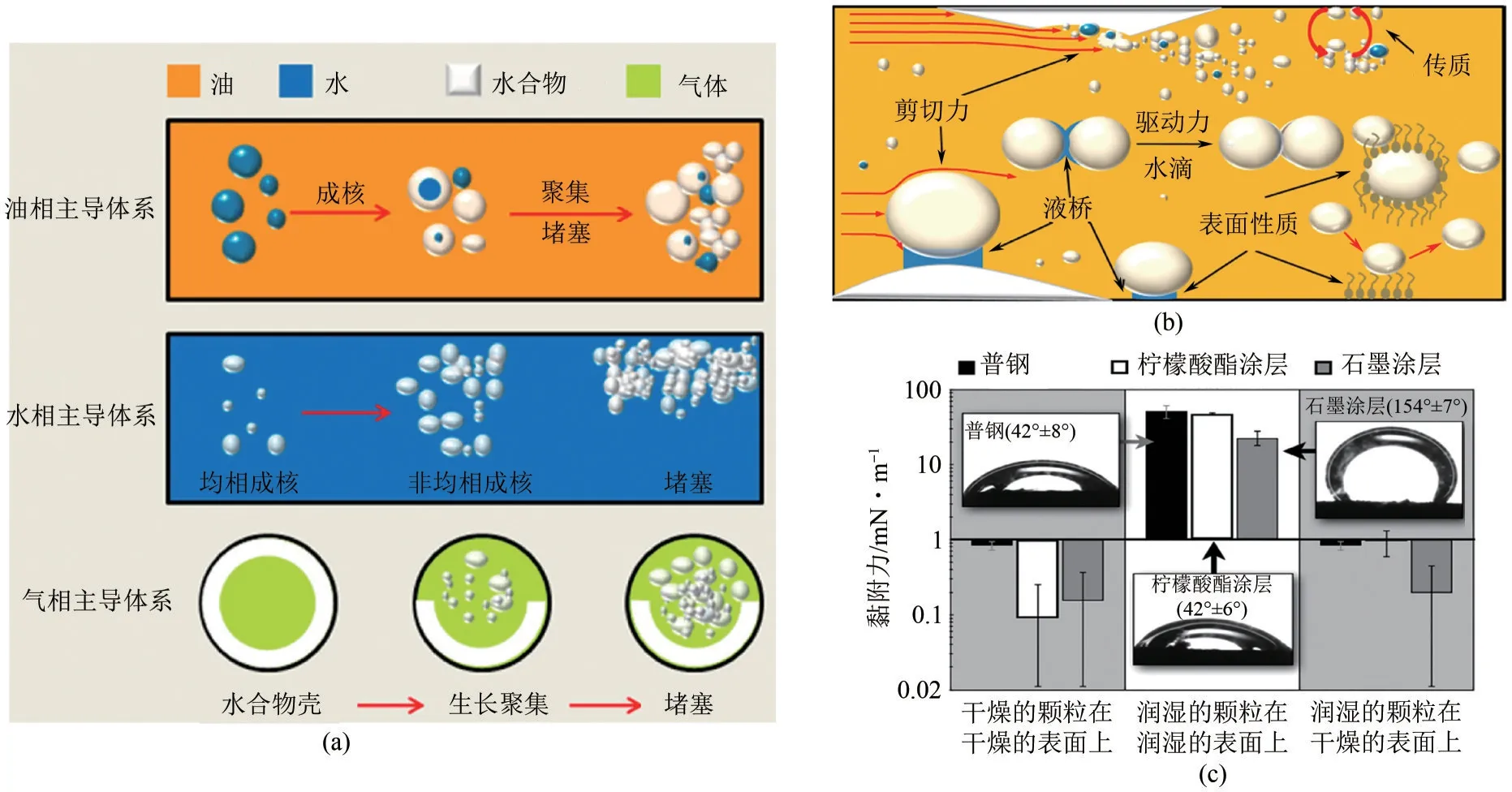
图4 水合物聚集沉积过程(a)[40]、影响因素(b)[41]以及亲疏水表面与环戊烷水合物间的黏附力(c)[42]
1.2.3 抑制水合物成核
目前传统防治水合物生成堵塞的化学方法主要有添加热力学抑制剂和动力学抑制剂[46-48]。热力学抑制剂(THIs)连续注入可以达到防止水合物生成的目的,这是因为THIs 可以改变水合物相平衡条件[49],使相平衡曲线迁移到较低的温度和较高的压力,使得水合物的生成在热力学上是不可行的,其可能的抑制作用机理主要有两种:①破坏水的亚稳态晶格。抑制剂大多为电解质,在水中电离产生的离子有较强的极性,减弱水分子之间的氢键作用,进而干扰水分子形成笼形结构。例如甲醇、氯化钙、盐等主要以这种方式起作用。②降低气相中水的蒸气压,从而不能满足生成水合物所需要的最低含水量。如乙二醇则属于这类抑制剂。
不同类型的热力学抑制剂抑制途径不同。醇类抑制剂是最成熟最常用的水合物成核抑制剂,例如甲醇、乙二醇等。醇的主要抑制机制是通过氢键与水分子相互作用来抑制成核。此外,醇存在条件下,水合物的生长过程中可能发生水合物晶格结构变形,这是由于水分子与醇的羟基和甲基之间的相互作用引起的晶格缺陷导致的。影响醇类抑制性能的因素,如图5(a)所示,醇类抑制剂的有效性取决于其结合水的能力,其通过氢键来强烈地结合水,强结合水与水合物成核竞争加剧,导致水与气体形成水合物的可能性较小。无机盐溶解在水中,其电离的离子通过静电力与水分子紧密结合,降低了水合物的分解温度,破坏了疏水性水合作用,增加了溶质吸附在水合物表面上的倾向,减少气体溶解到水相或界面处,进而抑制水合物形成。影响无机盐抑制性能的因素,如图5(a)所示,无机盐离子的水合物抑制强度取决于离子的浓度、电荷和半径。无机盐的浓度在其抑制性能中起着至关重要的作用,通常随着浓度的增加而增加,这是因为增加盐浓度,甲烷等客体分子引入水相中的自由能增加,降低了氢键的数量,扰动了水分子形成水合物所需氢键的形成,并且抑制水合物聚集。离子含量越高,半径越小,对水合物的抑制作用越好。离子半径在改变水合物平衡方面比离子电荷更重要。
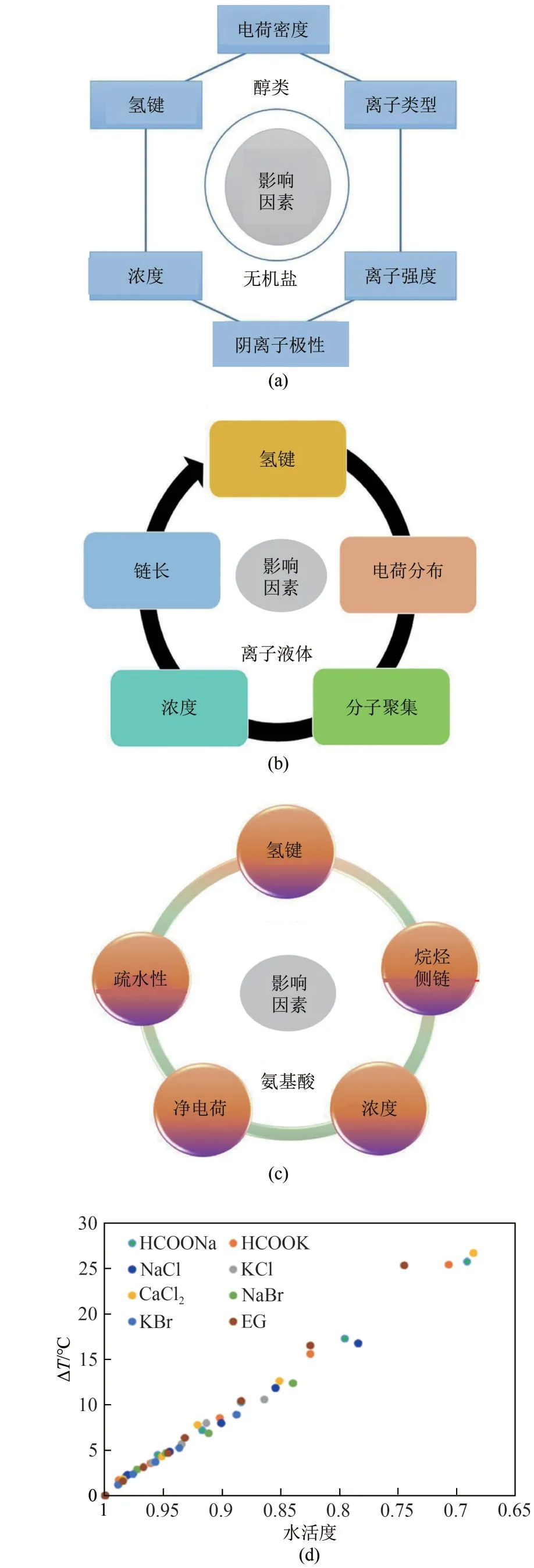
图5 影响不同抑制剂抑制性能的因素(a~c)]及其过冷度随水活度的变化(d)[50-51]
离子液体具有热力学和动力学抑制特性[52]。离子液体(ILs)亲水部分与水形成氢键网络并降低水合物的形成速率,同时将平衡水合物稳定性曲线转变为低温,最终诱导热力学抑制。溶解的离子通过水溶液中的静电力与水分子紧密结合,离子对水结构的影响取决于它们的电荷和尺寸。IL结构中能够形成氢键的官能团,将提供更有效的气体水合物抑制性能。影响离子液体抑制性能的因素,如图5(b)所示,ILs 热力学抑制性能取决于阳离子和阴离子大小、电荷大小和浓度(低浓度促进,高浓度抑制)。ILs吸附在增长的水合核表面,它们改变了晶体形貌或团簇表面的能量,从而影响水合物生长并最终延迟。目前,在所研究的所有离子液体中,四甲基氯化铵是最有效的热力学抑制剂。
氨基酸具有在水中形成两性离子的性质,通过它们形成氢键网络和静电相互作用,氨基酸的氢键增大了水分子重新定向排列的动力学扰动,氨基酸的静电吸引力可以影响水结构降低了水的活性,导致更大的抑制作用[51,53-54]。氨基酸产生的动力学抑制是由于侧链烷基对水的结构的扰动,水分子氢键网络的破坏显著延迟了水合物的形成。影响氨基酸抑制性能的因素,如图5(c)所示。氨基酸的抑制强度受到其通过与水分子的氢键结合而降低水合物形成中水活性的能力的影响,这取决于侧基烷基链,氨基酸的浓度和物理化学性质。Wang等[55]研究发现调节聚天冬酰胺结构可以有效延迟四氢呋喃(THF)水合物的形成,聚天冬氨酰胺的长侧链烷基可以导致水合物笼的变形,其亲水基团可以与水分子结合形成高度不可冻结的结合水并与THF水合物之间形成强烈的氢键。较长的烷基侧链导致水合物笼的更为有效变形并破坏笼形结构,对THF水合物具有更好的抑制效果。
Zhao等[50]研究表明,一定压力下,水合物抑制温度主要取决于水溶液的水活度,而与类型无关,并表现出线性关系。如图5(d)所示,包括无机盐、有机盐和乙二醇在内的8种THIs的水合物抑制作用随着水溶液中水活度的降低而增加。THIs 是高极性分子(盐、离子),其通过降低水的活性来抑制水合物的形成,THIs 的有效性取决于其结合水的能力。从根本上说,抑制水合物成核即抑制水合物笼形的形成,关键是破坏或者扰乱形成水合物笼的氢键形成,可以选择具有强结合水能力的官能团降低水的活性,抑制水合物的形成。
1.2.4 防止水合物黏附聚集
虽然热力学抑制剂可以永久性抑制成核,但是其具有用量大、回收困难、危害环境、运行成本高等问题,迫使研究转向一些低剂量动力学水合物抑制剂(LDHIs)的开发[56-57],如动力学水合物抑制剂(KHIs)和水合物防聚剂(AAs)。前者可以在水合物晶核达到临界尺寸之前,通过扰乱晶格结构和水分子簇的结构来抑制水合物成核。水合物成核之后,抑制剂分子通过氢键吸附在晶核表面,从而抑制水合物晶核的生长;后者则是通过阻止水合物颗粒聚集从而达到防止水合物堵塞的目的。动力学抑制剂通常是水溶性聚合物或共聚物,例如聚(N-乙烯基吡咯烷酮)以及绿色抑制剂AFPs 等。高效KHI一般具有酰胺基团和疏水官能团。KHI的活性基团、疏水基团的大小和形状、侧链的长度、疏基团之间的间距等也会影响KHIs 的性能。相比于THIs,KHIs 降低了用量和生产成本,但是过冷度过高(10℃以上)时,KHIs 溶解度降低抑制性能随之降低。虽然AAs 适用于更低温、高压的环境,但对于含水率大的体系,AAs容易失效,其性能受到油相组成和含盐量等因素的影响[58-60]。
相比于化学添加剂,开发防水合物黏附涂层更具优势。水合物在石油和天然气管道表面聚集生长导致管道堵塞,最小化水合物的体积和水合物与管道表面的黏附可以有效地解决这个问题。因此,深入了解影响水合物与管壁间黏附力大小的因素就非常重要。如图6(a)所示,Smith等[61]研究发现通过降低路易斯酸、路易斯碱和范德华力对表面自由能的贡献,实现表面与水合物黏附力最小化。水合物的黏附强度与实际黏附功相关,即具有低表面自由能的固体导致最低的水合物黏附力。Aman 等[62]通过控制表面粗糙度对矿物表面黏附强度进行量化,结果表明水的接触角对表面粗糙度具有非线性依赖性。如图6(b)~(d)所示,黏附强度也受到表面粗糙度和材料疏水性的显著影响,具有低粗糙度和强疏水性(即高接触角)的表面导致较低的黏附强度[63]。Aspenes 等[64]研究发现油气管道输送的组分中含有抑制水合物成核的成分,其中的石油酸可改变管道润湿性防止水合物聚集成大的水合物聚集体。当水滴润湿固体表面时,水合物与固体表面之间的黏附力是水合物-水合物黏附力的10 倍以上[图4(c)]。对于没有自由水的系统,系统中固体和水合物之间的黏附力太低而无法测量。这表明水合物黏附力大小强烈依赖于系统中水的存在,润湿管壁表面上的水合物沉积可能是水合物堵塞的主要途径。
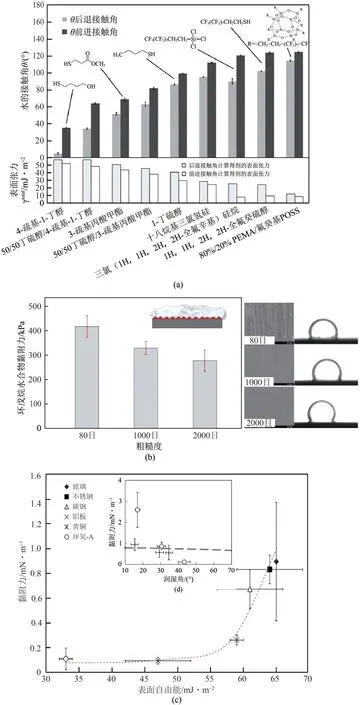
图6 材料表面能(a、c)、粗糙度(b)以及接触角(d)对水合物黏附力的影响[61,63-64]
不难看出,水的存在对水合物-水合物以及水合物-表面的黏附力影响最大,因此疏水是水合物防黏附的关键。除此之外,水合物与基材表面的附着力取决于材料表面性质,具有低表面自由能的固体表面导致较低的黏附力[64-66]。因此,通过调控表面微纳米结构和降低表面能,降低水合物的附着力,减少水合物与管壁的接触,对防止油气海底管线中水合物-基材表面沉积堵塞至关重要[67]。
1.3 特殊浸润材料与防水合物成核、黏附间的关系
特殊润湿性材料是新型仿生功能材料[68],其种类繁多[69-70],在抗结冰[71]、油水分离[72-74]、气体液体运输[75-76]、电化学等领域有着广泛应用[77-78],这归因于其特殊的界面性质。特殊润湿性材料的各种特性通常是由微纳米结构与表面能协同作用的结果。Smith等[61]研究表明与纯钢相比,水合物-疏水表面THF水合物的黏合强度降低了四倍以上,为此提出通过降低表面自由能实现水合物黏附功最小化来实现。此外,纳微米级表面形态和化学性质可以进一步降低水合物的附着力,防止水合物渗透到纹理基材表面的微观粗糙结构中,促进Cassie-Baxter状态的建立,使得水合物在基材表面具有非常高的后退接触角。通过分析极性和非极性流体的前进和后退接触角对表面能进行量化,后退接触角测量可以作为预测水合物黏附强度和快速筛选水合物疏水涂层简单、有效的工具。低表面能的特殊浸润材料会影响基材与水合物、客体分子以及水分子簇间的表面张力、界面间的相互作用,同时其微纳米结构将会影响界面接触面积、界面处客体分子浓度分布。这些因素对特殊浸润性材料在水合物成核与聚集领域中的应用至关重要,接下来将围绕对特殊浸润材料在水合物形成与抑制、聚集与沉积方面的展开研究。
2 特殊浸润性材料对水合物成核、聚集沉积的影响
2.1 亲疏水性对客体浓度以及水分子有序性的影响
若将水合物成核类比化学反应,影响其化学反应的因素,除了温度、压力,还包括反应物的浓度(主客体分子的浓度)、催化剂[化学添加剂(表面活性剂等)、固体相(纳米粒子等)],如图7(a)所示,客体分子浓度越大成核时间越短[79]。在疏水水合作用下,疏水官能团或疏水表面均会影响主客体分子浓度分布的影响。Farhang 等[80]提出存在于水和疏水表面界面处的致密气体层促进天然气水合物的形成。因为在气-水界面处的致密气体层局部摩尔分数可以非常高。在疏水表面存在高气体密度,这可以提供足够量的气体用于水合物成核和生长。如图7(b)所示,Nguyen等[81]通过模拟和实验结果结合的方式证明了疏水界面富集(IGE)的存在以及疏水表面上更多水的局部排序的趋势,导致在含疏水颗粒的体系中促进气体水合物的形成。如图7(c)所示,疏水固体颗粒在促进天然气水合物形成过程中发挥着关键作用[82],如图7(d)所示,随疏水性增大水合物成核诱导时间成非线性降低[83]。此外,多数研究人员认为是纳米颗粒增加的接触面积,提供了更多异质成核位点,如图7(e)所示,腐蚀后的表面接触面积增大诱导时间显著降低[84],忽略了疏水性表面能够富集气体,强化气液传质,进而促进水合物成核。
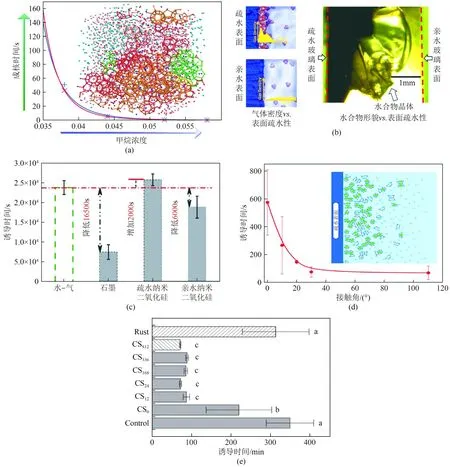
图7 亲疏水表面对水合物成核时间(a、c)[79]、客体分子浓度分布(b)[81]的影响以及材料接触角(d)[83]和粗糙度(e)[84]对诱导时间的影响
疏水表面或者疏水官能团不仅可以通过富集气体促进水合物成核,还可以诱导水分子有序排列,降低成核垒,进而促进水合物成核。如图8(a)所示,Deng等[85]分子模拟结果表明,随官能团疏水性的增大,水分子趋于有序。此外,液体分子有序结构对晶体成核和生长具有调节作用[86]。疏水官能团不仅能够将周围的水分子组织成笼形结构,而且能够增加疏水官能团周围气体浓度,从而促进水合物的形成。相对应地,亲水官能团会破坏周围的水结构并与气体竞争水分子抑制水合物的形成[81]。疏水性较低的氨基酸(具有较小的烃基或较强的亲水基团)强烈破坏局部水结构并有效地抑制气体水合物的形成。相反,具有较高疏水性的氨基酸增强了局部水结构并起到水合物促进剂的作用。特别地,当添加剂是两亲性添加剂时,观察到的效果是疏水部分(促进剂)和亲水部分(抑制剂)之间竞争的结果[87]。此外,如图8(b)所示,Park 等[88]研究发现二氧化碳溶解之后施加电场气体水合物成核速度明显加快,诱导时间缩短了5.8 倍,这主要归因于施加电场会产生冰状有序结构并将扩散系数降低大约1个数量级。有趣的是,在二氧化碳溶解之前向水施加外部电场时,水-气界面处存在强极化水层会阻止气体吸收,从而抑制气体水合物成核;Nguyen等[89]发现了类似的现象,十二烷基硫酸钠(SDS)已被证明可以强烈促进甲烷水合物的形成,当表面活性剂的浓度低于毫摩尔浓度(约0.3mmol/L)时,SDS 对甲烷水合物的形成显示出优异的抑制作用,如图8(c)所示,当添加带相反电荷的表面活性剂(四正丁基溴化铵,TBAB)时,水的有序和抑制效果都消失了,这归因于TBAB的使用减轻了SDS在溶液表面产生的静电场,因此削弱了界面水的排列,从而消除了抑制作用[90-91]。Wang等[83]采用十八烷基三氯硅烷(OTS)涂覆砂粒表面使其疏水化,更容易形成甲烷水合物,并且随着砂粒表面疏水性的增加,水接触角的增加,诱导时间稳定地减少,这可能是由于疏水表面增加水分子有序排列促进成核。如图8(d)所示,Li等[90]原位拉曼光谱结果表明,相比在液相中、亲水或部分疏水附近,在疏水表面附近水分子更容易形成冰状有排序。综上,疏水表面或疏水官能团可以通过富集气体以及诱导水分子有序排列促进成核,亲水则相反。水分子有序排列可以降低成核能垒,促进水合物成核,但是高度有序排列的水分子通过阻碍气液传质的方式,宏观表现为抑制水合物成核。不难推断,对于水溶性客体分子或者过饱和非极性客体分子,疏水表面或疏水官能团附近水分子有序排列将会促进水合物成核,亲水表面或者亲水性官能团则会抑制成核。需要说明的是,不是亲水就可以抑制水合物成核[92-94],能够与水分子氢键结合,形成强氢键官能团是必须的。
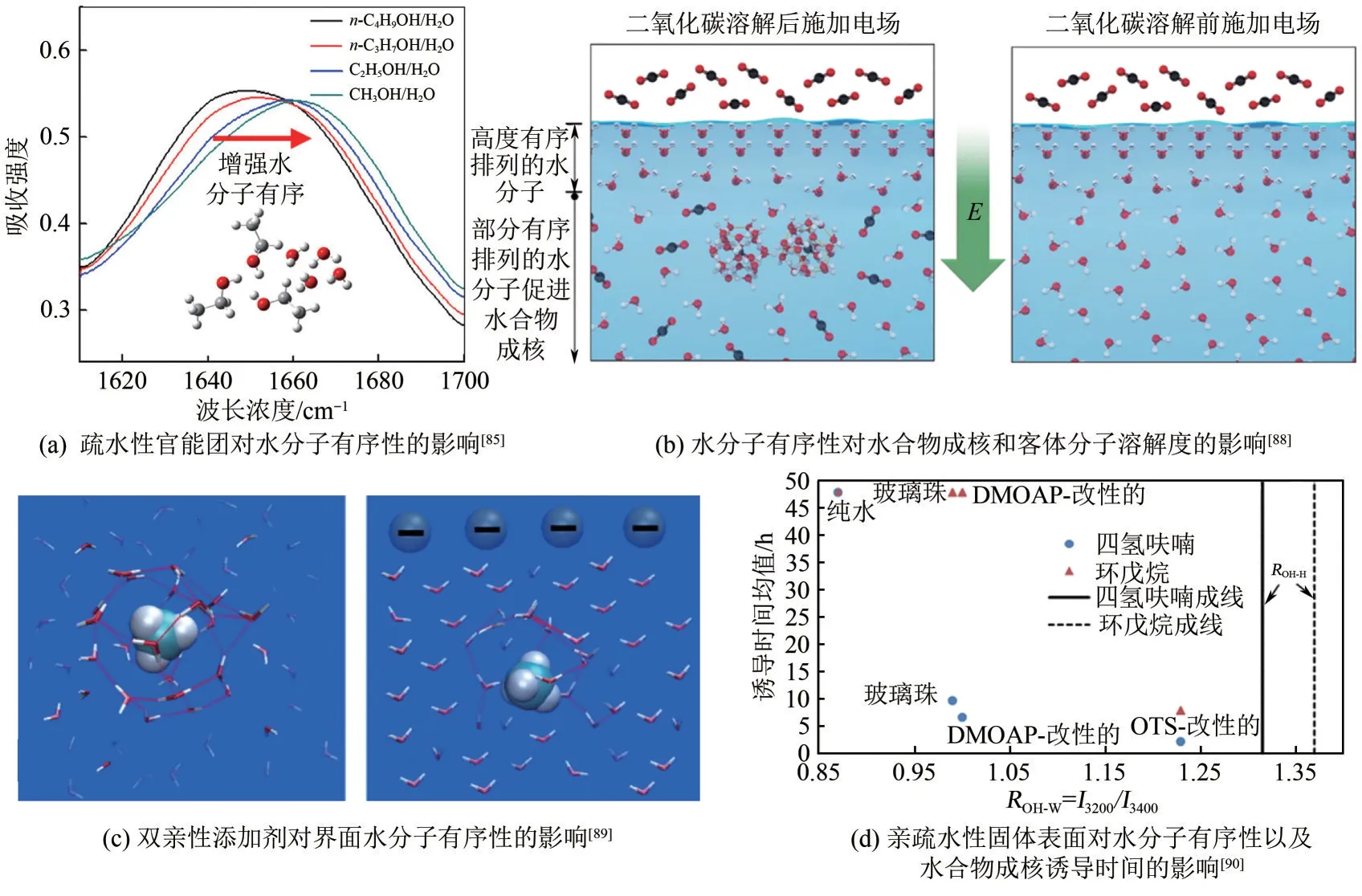
图8 水分子有序性对水合物成核的影响
2.2 亲疏水材料对水合物成核的影响
研究人员比较了不同浸润性颗粒对诱导时间的影响,结果表明固体颗粒表面浸润性在天然气水合物成核过程中发挥着重要作用[15,37,95-96]。Park等[97]研究表明疏水性固体也可以作为热力学促进剂,疏水二氧化硅可以使甲烷气体水合物的平衡条件转变为较低的压力和较高的温度。He 等[98]通过分子动力学模拟发现疏水性石墨具有吸附CH4分子并促进CH4引入水中的能力,CH4分子吸附在石墨表面上,并且在本体区域发生水合物成核;此外,疏水性二氧化硅粉末、干水对二氧化碳和甲烷水合物都有极好的促进作用[80,99-100]。Saha 等[101]多孔介质(孔径49Å)将THF-H2二元笼形水合物从在冰体系中的形成时间3~10h 减少至到27min。笼形水合物形成时间随着多孔介质的孔径而增加,氢气扩散率随着孔径或笼形水合物粒径的增加而降低。Liu 等[102]研究表明多孔介质的粒径越小,水合物形成过程的诱导期越短,复合体系中形成的水合物的气体吸收量越大。Casco 等[103]研究了在2℃和3~5MPa 条件下金属有机骨架(MOF)材料上甲烷水合物的形成。研究发现疏水性MOF 能够促进甲烷水合物形成,但亲水性MOF 不能,可能是由于亲水孔腔内水的存在极大地抑制了甲烷的吸收,而疏水性MOF 可以抑制水分进入内部孔隙,因而在MOF 外部形成水合物。Kim等[104]研究发现纳米尺寸的空间和氧化石墨的官能团亲水表面(羧基、羟基等)使得在氧化石墨中形成的天然气水合物成核受到强烈抑制。Wang等[91]研究表明与超纯水相比,亲水性纳米SiO2流体将水合物形成的诱导时间增加了194%,并将水合物形成的量和平均速率分别降低了10%和17%。更重要的是,Hall 等[105]证明了多元醇涂层(APTES SA IG)抑制四氢呋喃水合物形成的可行性。
研究人员通过对传统多孔材料浸润性能改性[32,106-115],这将极大的丰富促进或抑制水合物成核材料可选择性。需要明确的是,不是将亲水抑制官能团接枝到基材表面就可以抑制水合物成核[116-117],疏水性固体表面也不一定促进成核[57,94,105,118-119]。例如,在油-水体系中,疏水性固体表面[15,104,120]、疏水性水溶性聚合物、天然氨基酸等将会抑制水合物成核[121]。综上,除温度压力、主客分子浓度等因素会影响诱导时间外,不同亲疏水性介质的加入对主客体分子分布、水分子有序性具有不同程度的影响。疏水性基材和气体之间的协同疏水相互作用导致疏水基材周围局部气体浓度增加。从热力学角度分析,疏水性固-水界面处的水分子具有增加局部有序性的趋势,而亲水性固-水界面的水分子显示出具有抑制水分子有序性排列,上述研究为制备抑制水合物成核材料提供了理论依据。
2.3 亲疏水材料对防水合物黏附的影响
近年来,越来越多的研究人员将特殊润湿性材料的应用拓展到油气储运,如图9(a)、(b)所示。涂层表面的疏水性越高,表面能越低,水滴在固体表面上的接触角越高,则水合物与固体表面间的黏附力越低[42,61,63-64,122]。Aman 等[42]由低表面能物质修饰微纳米结构制备的超疏水表面,因具有更高的接触角和更小滚动角,在水合物防黏附方面表现巨大的应用潜力。Sojoudi 等[123-124]通过十八烷基三氯硅烷(OTS)处理的超疏水表面黏附力小于0.004mN/m;制备的双层聚二乙烯基苯/聚(全氟癸酸丙烯酸酯)涂层,前进接触角[(148.3±4.5)°]和退后接触角[(142.5±9.8)°],其与水合物黏附强度从(220±45)kPa降低至(20±17)kPa,结果表明具有较高氟浓度的低表面能表面显著THF 水合物的黏合强度。Dong等[125]通过硬脂酸修饰电沉积氧化铜得到超疏水表面,其与水合物附着力为0.00013N/m,远低X90管线钢表面之间的附着力(0.0016N/m)。Das 等[126]通过促进水合物和固体表面之间的环戊烷形成液膜来降低环戊烷水合物的黏附性。如图9(c)所示,纯硅表面能(53mJ/m2)表现出最大水合物积累量,两种硅烷[十八烷基三氯硅烷(OTS) 和十三烷基-1,1,2,2四氢辛基-三氯硅烷(FS)]的改性表面基本无水合物形成,与其低表面能相一致。然而,具有最低表面能(8mJ/m2)的FS 涂层表面与具有更高表面能(24mJ/m2)的OTS 涂层表面相比具有更大的水合物晶体覆盖率。原因在于OTS涂层接触角滞后、滚动角更小以及涂层的非极性基团形成的液膜限制了水和固体的接触面积。更多超疏水材料用于防水合物黏附见表1。

表1 超疏水材料防环戊烷水合物黏附汇总
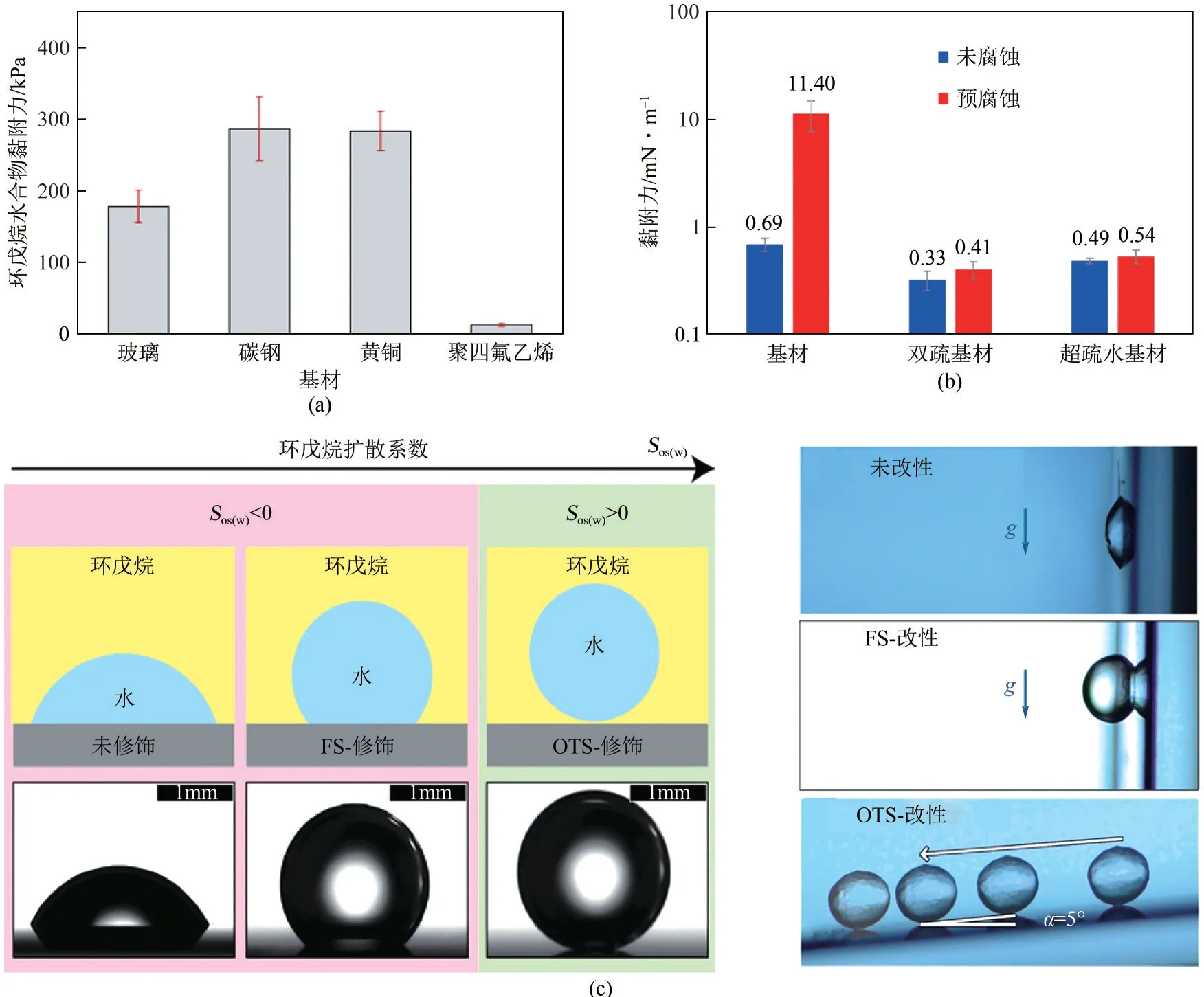
图9 亲疏水材料表面(a、b)[63,122]和固体表面扩散系数Sos(w)(c)[126]对水合物黏附力的影响
显然,超疏水材料将有助于减小水合物在基材表面黏附性,对减缓甚至避免水合物集聚沉积具有潜在应用价值。这主要归功于超疏水表面的微纳米结构减少了固-液接触面积,其低表面能极大降低黏附力,进而可以有效降低水合物与管壁间的黏附力。
3 特殊浸润性材料应用技术难点及解决设想
对于天然气主导运输体系,亲水表面水合物黏附力是疏水表面黏附力十倍以上,似乎高接触角低表面能超疏水或超双疏材料是防止水合物黏附的理想材料。但是上述材料仅限于防止已经成核的水合物黏附于管壁,忽视了疏水表面容易诱导水分子疏水化[131-133],相比亲水表面,疏水表面水分子排列更加有序,降低了水合物成核能垒,促进水合物笼形结构的形成[85,90]。例如,Aspenes等[64]研究发现柠檬酸酯涂层使水合物黏附力降低98%的同时也加速了水合物颗粒的生长。这也反应了疏水表面的两面性,即可以极大程度的降低已经成核的水合物与管壁间的黏附力,但也存在促进新的水合物成核生长的风险。最近,Yin等[134]通过分子模拟和实验证实并制备了一种具有良好的机械性能和化学稳定的含羟基超疏水材料,该材料不仅可以抑制水合物成核还可以防水合物黏附,这为制备多功能防水合物成核黏附材料的制备提供新的思路。但是研究体系是客体分子与水分子互溶体系,材料是否适用于气相主导的体系,例如甲烷-水体系还需进一步验证。在气相主导的运输体系中,在超疏水材料表面微纳米结构形成的空气层,其主要成分是甲烷,甲烷气体会在高比表面积的微纳米结构富集[135],这可能会促进水合物异质成核,一旦成核,超疏水表面就丧失防水合物黏附功能。因此,超疏水材料应用在气相主导的油气管道是有促进水合物成核的风险。若在微纳米凹槽能形成稳定的惰性气囊,上述问题将迎刃而解。Hu 等[136]首次证明,通过用交替的超疏水和亲水圆周带覆盖Taylor-Couette 流动装置的内转子,可以形成大的表面能垒以强有力地固定空气/水/固体的三相接触线,这可以防止空气层的破坏,形成稳定且连续的空气环。
对于油相主导的运输体系,本文作者认为大多数超疏水材料是无法防水合物黏附的。原因在于防水合物黏附测试环境多是环戊烷为连续相的体系,忽视了实际运输介质包含原油,高黏度原油很容易污染超疏水材料表面,导致超疏水材料丧失防水合物黏附性能。超双疏材料虽然可以防原油黏度,但是也会富集气体,促进水合物成核[137]。此外,深海管道温度较低,冷凝油滴可能会润湿超双疏材料表面微纳米结构,导致超双疏性能丧失,失去防水合物黏附性能。因此防水滴、油滴冷凝是制备防止原油、水合物黏附聚集超双疏材料的必要条件。研究表明分级微纳米结构拓扑化纳米草[138]、不规则凹入式微腔和纳米结构组成的超疏水铝材[139]、含有高孔径比MWCNTs[140-141]以及硅纳米管[142]超疏水涂层显示出优异的防冷凝性能[143]。除此之外,具有三维高孔隙率的高孔径比基材更有助于实现更好防冷凝性能[144-147]。Liu 等[148]制备了由底层半互穿阵列结构和表层微纤维网络结构组成的超疏水表面。与单阵列TiO2纳米棒相比,紧凑的半互穿阵列结构可以有效防止水蒸气的扩散和冷凝完全润湿表面,因为TiO2纳米棒阵列之间的聚合物网络隔离层具有良好的抑制作用。基于上述研究,可以归纳得出通过调控材料表面纳米孔(纳米阵列)的柱高度与间距比值[143,147,149-153],经由低表面能物质修饰后制备具有防水滴、油滴冷凝的超双疏材料,将会是防止原油、水合物黏附聚集堵塞管道有效途径。
对于水相主导的运输体系,抑制剂性能通常会受到高含水率以及原油等组分干扰的影响。超双疏材料可以疏水、疏原油,但是无法抑制水合物成核,反而其微纳米结构可能会束缚客体分子,促进水合物异质成核[137]。超亲水材料能够与水分子结合形成水合层,从而防止原油黏附和污染[154-155],但是未必能够抑制水合物成核。Hall等[105]证明了多元醇涂层抑制四氢呋喃水合物形成的可行性。这种表面接枝抑制官能团的方法为制备抑制水合物成核材料提供新的思路。但是二维材料表面抑制官能团接枝密度有限,而抑制性能与抑制官能团浓度相关,通常抑制剂浓度越大抑制性能越强。具有高比表面积的三维微纳米颗粒以及三维多孔介质(泡沫铝,泡沫铜,开孔碳化硅等)[20-21,156]能够提供更多异质成核位点,进而更容易促进水合物成核。若将上述具有亲水抑制涂层涂覆到具有高比表面积的三维多孔材料表面,或许可以实现促进水合物成核的材料转为抑制成核的材料[134],同时解决了使用化学抑制剂或者改性纳米粒子等方法带来的分离回收、污染环境等问题。
4 结语
综上,依据抑制官能团抑制水合物成核机理,对特殊润湿性材料的微纳米结构和表面化学性质进行设计和调控,制备了具有不同功能的特殊润湿材料,是以应对不同运输介质体系中水合物成核聚集堵塞管道的有效途径。制备具有抑制水合物成核性能的特殊润湿性材料,首先是其在高比表面积表面接枝抑制官能团,破坏形成水合物笼形结构所需的水分子间氢键,即弱化分子间氢键稳定性,与水分子氢键结合扰乱水分子有序排列形成冰簇或水合物笼;其次,减少气-液接触面积,避免客体分子在界面处富集以及提供异质成核位点;防止水合物聚集沉积的特殊润湿性材料制备关键是避免材料表面被水润湿,水的存在极大增加了界面之间黏附力。超双疏材料可以避免因水润湿导致的黏附力急剧增大,也可以防止高黏度原油黏附污染,极大降低固-液、固-固接触面积,其低表面能进一步降低水合物与材料表面间的黏附力,因此超双疏材料是降低水合物黏附力的理想材料。尽管特殊浸润材料为抑制水合物成核堵塞注入新的活力,但对于特殊浸润材料在水合物成核领域的应用研究较少,缺少针对不同运输介质体系的系统性研究。考虑到油气管道苛刻环境要求,具有耐酸碱腐蚀、耐高压、耐磨损、防低温冷凝,同时可以抑制水合物成核和防水合物黏附材料的制备,在未来将会是研究的热点与重点。
猜你喜欢
西南石油大学学报(自然科学版)(2021年3期)2021-07-16 05:27:08
科教新报(2021年11期)2021-05-12 19:50:11
现代纺织技术(2019年2期)2019-09-10 07:22:44
烟草科技(2019年2期)2019-02-23 07:15:46
西南石油大学学报(自然科学版)(2018年6期)2018-12-26 01:00:14
现代纺织技术(2018年4期)2018-09-10 05:45:05
中国资源综合利用(2017年4期)2018-01-22 02:46:57
河北地质(2017年2期)2017-08-16 03:17:10
科学之谜(2016年9期)2016-10-11 08:59:04
中国环境科学(2016年2期)2016-04-16 07:12:16
