
FOXQ1 敲除乳腺癌MCF7 细胞株的构建及其功能初探
2023-09-28 07:45杨欢冯玉梅
天津医科大学学报 2023年5期
杨欢,冯玉梅
(天津医科大学肿瘤医院肿瘤研究所生物化学与分子生物学研究室;国家肿瘤临床医学研究中心;天津市“肿瘤防治”重点实验室;天津市恶性肿瘤临床医学研究中心;乳腺癌防治教育部重点实验室,天津 300060)
缺氧微环境是实体瘤的普遍特征。Voss 等[1]研究发现,缺氧环境可导致乳腺癌细胞迁移能力的增加。此外,多个研究表明,原发性肿瘤氧合不良的患者转移率和死亡率增加[2-4]。缺氧诱导因子1α(hypoxia-inducible factor 1α,HIF-1α)是体内多器官系统中细胞缺氧反应的主要转录因子和调节因子[5]。在常氧环境中HIF-1α 的脯氨酸残基发生羟基化使其被泛素化降解,而缺氧环境中HIF-1α 脯氨酸不再发生羟基化,从而导致HIF-1α 蛋白的积累[6]。
叉头框(forkhead box,FOX)蛋白是在进化上保守的转录因子超家族,在癌症的发生、发展中发挥重要作用[7]。许多FOX 蛋白被报道直接或间接响应缺氧信号而被激活表达[8-11]。FOXQ1 是FOX 转录因子家族成员之一,其在包括乳腺癌在内的多个癌种中被表征为上皮间质转化(epithelial-mesenchymal transition,EMT)的主要激活剂,赋予细胞转移和侵袭的能力[12-15];并且其在间充质基质细胞(mesenchymal stromal cells,MSCs)中被报道可能间接响应于缺氧信号而表达上调[11]。因此推测,在乳腺癌中,处于缺氧微环境的癌细胞其可能响应于缺氧信号导致FOXQ1 表达上调,从而产生具备较高转移或侵袭能力的亚克隆。
为了构建有效的实验模型,选择通过成簇的规则间隔短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)/CRISPR相关蛋白9(CRISPR-associated protein 9,Cas9)系统来构建FOXQ1 基因敲除的MCF7 细胞株。该系统由Cas9 蛋白、CRISPR RNA(crRNA)和反式激活CRISPR RNA(trans-activating crRNA,tracrRNA)组成。其中,crRNA 作为识别模块能够识别相应的特异性靶序列;tracrRNA 和crRNA 的核苷酸链能互补配对构成单指导RNA(single guide RNA,sgRNA),sgRNA 与Cas9 一同形成核糖核蛋白复合物(ribonucleoprotein,RNP)并同时激活Cas9 作为核酸内切酶的功能[16-17];Cas9 切断目的区域的双链核酸,诱导细胞内源DNA 损伤修复机制工作,最终借助该机制达到编辑遗传信息的目的[18]。
1 材料与方法
1.1 实验材料 人乳腺癌细胞系MCF7 来自American Type Culture Collection;DMEM 高糖培养基、胎牛血清和青霉素/链霉素购于美国Gibco 公司;Leti-CAS9-puro 单载体慢病毒购于吉凯基因;crRNA 和tracrRNA 由广州锐博生物合成;Lipofectamine 3000转染试剂购于美国Invitrogen;T7 核酸内切酶I(T7 Endonuclease I,T7E1)和NE Buffer 购于美国NEB 公司;DNA 提取试剂盒、DNA 纯化试剂盒、PrimeSTAR Max DNA Polymerase 均购于日本Takara 公司;FOXQ1 抗体购于美国Santa 公司;HIF-1α 抗体购于美国CST 公司;β-Actin 抗体购于武汉ABclonal 公司;transwell 购于美国Corning 公司;结晶紫染液购于上海碧云天公司。
1.2 实验方法
1.2.1 crRNA 设计合成 从NCBI 中检索人FOXQ1 基因的外显子序列,根据Cas9 靶点设计原则,即待编辑的区域附近存在相对保守的PAM 序列(NGG),并针对其外显子碱基序列设计3 个敲除位点(表1)。根据敲除位点设计合成相应的crRNA以及与之配套的tracrRNA,KO#1、KO#2 和KO#3 3个敲除位点对应的crRNA 分别命名为FOXQ1-crRNA#1、FOXQ1-crRNA#2 和FOXQ1-crRNA#3。

表1 敲除位点识别序列Tab 1 Knockout site recognition sequences
1.2.2 构建Cas9 工具细胞株 将1×105个MCF7细胞接种于24 孔板,待细胞融合度达到约30%时,加入2 μL Leti-CAS9-puro 慢病毒。细胞感染12 h观察细胞状态,如果没有明显的细胞毒性作用,继续培养24 h 后弃去含有病毒的培养基,更换为新鲜DMEM 完全培养基。感染2 d 后更换培养基为含2 μg/mL 嘌呤霉素的DMEM 完全培养基进行连续3 d 的药物筛选,由此获得Cas9 稳定表达MCF7细胞株,作为MCF7-Cas9 工具细胞待用。
1.2.3 转染FOXQ1-crRNA+tracrRNA 接种1×105个MCF7-Cas9 工具细胞于24 孔板,待细胞融合度达到50%~70%时进行FOXQ1-crRNA+tracrRNA的转染。取一支1.5 mL EP 管,加入25 μL Opti-MEM减血清培养基,再分别加入2 μL FOXQ1-crRNA和EP 管,加入25 μL Opti-MEM 减血清培养基,再加入3 μL lipo3000 转染试剂;而后将FOXQ1-cr-RNA+tracrRNA、Opti-MEM 混合物加入到Opti-MEM 减血清培养基稀释的lipo3000 转染试剂中,室温孵育5 min。孵育结束后,将上述制备好的转染混合物加入到细胞培养基中,于37℃、5%CO2条件下培养,48 h 后更换新鲜DMEM 完全培养基,并继续扩增培养,供后续实验使用。
1.2.4 T7EI 酶切检测靶位点编辑效率 目的基因片段经PCR 扩增后重新变性退火变成异源双链DNA,T7EI 酶能够识别不完全匹配的双链DNA,从不匹配的位置将其酶切,利用这个性质能够鉴定CRISPR/Cas9 编辑形成的突变体,评估crRNA 的编辑效率。转染FOXQ1-crRNA+tracrRNA 的MCF7-Cas9 细胞扩至12 孔板后,取一半细胞按试剂盒步骤提取基因组DNA,并用微量紫外分光光度计定量。PCR:吸取50 ng 基因组DNA 用PrimeSTAR Max DNA Polymerase 进行PCR 扩增,扩增出含crRNA靶序列的片段,其引物信息见表2;PCR 产物用DNA纯化试剂盒进行纯化,并用微量紫外分光光度计进行定量。PCR 产物变性退火:取200 ng 纯化后的PCR产物,加入2 μL NE Buffer,并补充无核酸水至19 μL,并根据T7E1 试剂盒提供的变温条件进行变性退火。T7EI 酶切:向退火后的PCR 产物加入1 μL T7EI酶,于37℃孵育15 min,加入1 μL 的proteinase K,37℃温育5 min 终止酶切。2%的琼脂糖凝胶电泳检测退火后的PCR 产物的酶切效率,酶切率越高则细胞靶位点编辑效率越高。

表2 引物序列Tab 2 Primer sequences
1.2.5 FOXQ1 基因敲除单克隆细胞株的筛选 用1.2.4 中筛选出的靶位点编辑效率较高的细胞消化后计数,计数后按1 cell/孔的量接种按至96 孔板进行培养,12 h 内于镜下观察,舍去多细胞孔,单细胞孔继续扩增培养;待单克隆细胞扩至12 孔板取一半细胞提取基因组DNA,再次用T7EI 酶鉴定单克隆细胞靶基因的编辑状态,实验方法同1.2.4。将有酶切效率的PCR 产物送至生工生物工程公司进行Sanger 测序,从DNA 水平鉴定FOXQ1 的敲除情况;根据DNA 测序结果筛选出FOXQ1 基因稳定敲除的MCF7 单克隆细胞株。而后,通过Western 印迹从蛋白水平进一步鉴定FOXQ1 的敲除。
1.2.6 Western 印迹 将细胞种于6 孔板,待细胞长满后加入适量细胞裂解液提取总蛋白,用BCA 法测定细胞蛋白浓度;各取40 μg 蛋白,用10%的SDS-PAGE 凝胶进行电泳,80 V 30 min,100 V 90 min;转膜,80 V 120 min;5%脱脂牛奶室温封闭1 h;分别加入一抗FOXQ1(1∶1 000)、HIF1-α(1∶1 000)和β-Actin(1∶5 000)4℃孵育过夜;次日TBST洗膜后加入对应的二抗(1∶2 000),室温孵育1 h后TBST洗膜,加入ECL 试剂化学显色。
1.2.7 划痕实验 将MCF7-FOXQ1-/-1 和MCF7-FOXQ1-/+2 及其野生对照MCF7-WT 细胞均匀接种于6 孔板中,待细胞融合度达到100%,用枪头在每个孔内划出2×2 横竖交叉的4 条线,PBS 清洗脱落细胞,更换新鲜DMEM 培养基,镜下拍照,记为0 h;常氧组和缺氧组分别置于20%O2和1%O2条件下培养,往后每12 h 拍照记录一次,并测量划痕间距;划痕愈合率=(0 h 划痕间距-培养后划痕间距)/0 h划痕间距×100%。
1.2.8 transwell 实验 于24 孔板中加入750 μL 含20%血清的DMEM 培养基,并将transwell 小室置于孔中;分别设置常氧(20%O2)和缺氧(1%O2)组,每组取生长状态良好的MCF7-FOXQ1-/-1 和MCF7-FOXQ1-/+2 细胞及其野生对照MCF7-WT 细胞各5×104个,重悬于500 μL DMEM 培养基中并接种于小室内,每个细胞设3 个重复。48 h 后取出小室,用棉签轻轻擦拭小室内部去除未发生迁移的细胞,4%多聚甲醛室温固定30 min,结晶紫室温染色30 min,PBS 清洗后置于室温风干。显微镜观察染色细胞,随机选取5 个视野拍照并统计细胞数。
1.2.9 生物信息学分析 从METABRIC 数据库中下载并整理了1 980 例乳腺癌患者的RNA-seq 数据,用于分析FOXQ1 与缺氧相关基因的表达相关性。
1.3 统计学处理 采用GraphPad Prism 8.0 软件进行数据分析和作图。数据符合正态分布,采用单因素方差分析和t 检验进行差异分析,采用Pearson 相关性分析评估基因间mRNA 表达的相关性。P<0.05 表示差异有统计学意义。
2 结果
2.1 FOXQ1 基因敲除单克隆细胞株的获取 转染FOXQ1-crRNA+tracrRNA 的MCF7-Cas9 细 胞 经T7E1 酶切检测出FOXQ1-crRNA#3 对细胞靶位点有最高的编辑效率。用该细胞进行单克隆培养后经T7E1 酶切验证出4 株有靶基因编辑效率的单克隆细胞(图1A)。通过进一步比对sanger 测序结果,从4 株有FOXQ1 基因编辑效率的单克隆细胞中鉴定出一株纯合子细胞和3 株杂合子细胞,依次命 名 为 MCF7 -FOXQ1-/-1、MCF7 -FOXQ1-/+2、MCF7-FOXQ1-/+3 和MCF7-FOXQ1-/+4。纯合子细胞株测序结果显示,与野生型MCF7 相比,其缺失了包括FOXQ1-crRNA#3 识别区域在内的57 个碱基,并在缺失序列后的第10 个碱基位点发生G-A 碱基突变(图1B)。

图1 FOXQ1 基因敲除单克隆细胞株的获取Fig 1 Acquisition of FOXQ1 gene knock out monoclonal cell lines
2.2 FOXQ1 基因敲除细胞株的蛋白表达验证 尽管基因移码突变对翻译过程有极大影响,但此4 株FOXQ1 敲除的细胞株中FOXQ1 蛋白的表达是否确切受到影响还需进一步验证。Western 印迹结果显示,与野生型MCF7-WT 细胞相比,纯合子MCF7-FOXQ1-/-1 和杂合子MCF7-FOXQ1-/+2 细胞株已完全不表达FOXQ1 蛋白,而杂合子MCF7-FOXQ1-/+3和MCF7-FOXQ1-/+4 还能检测到一部分FOXQ1 蛋白的表达。理论上,基因敲除的杂合子细胞对基因的编辑效率应为50%,但FOXQ1在MCF7 细胞中本身是低表达或不表达的,因此MCF7-FOXQ1-/+2 可能是一个FOXQ1 本底表达足够低的单克隆。所以,成功构建了两个FOXQ1 基因稳定敲除的MCF7 细胞,即纯合子MCF7-FOXQ1-/-1 和杂合子MCF7-FOXQ1-/+2 细胞株,故后续实验选取此两株进行(图2)。

图2 FOXQ1 基因敲除细胞株的蛋白表达验证Fig 2 Verification of protein expression in FOXQ1 knockout cell lines
2.3 缺氧促进MCF7 细胞中FOXQ1 的表达 Western印迹结果显示,与常氧相比,缺氧环境中,MCF7-WT细胞FOXQ1 蛋白表达显著上调。相应的,FOXQ1 敲除杂合子细胞MCF7-FOXQ1-/+2 也显示出部分FOXQ1 蛋白的上调(图3)。

图3 缺氧促进MCF7 细胞中FOXQ1 的表达Fig 3 Hypoxia promotes FOXQ1 expression in MCF7 cells
2.4 FOXQ1 介导缺氧引起的MCF7 细胞迁移能力的增加 划痕实验和transwell 实验结果显示,与常氧相比,缺氧显著上调MCF7-WT 的转移能力(t=3.78,P<0.01;t=11.94,P<0.001),而FOXQ1 敲除的纯合子细胞MCF7-FOXQ1-/-1 在缺氧条件下的转移能力基本与常氧条件下持平(t=0.63,P=0.55;t=2.54,P=0.06)。相应的,FOXQ1 敲除的杂合子细胞MCF7-FOXQ1-/+2 其在缺氧条件下的转移能力有一定程度的上调(t=2.46,P<0.05;t=4.95,P<0.05),但其上调程度远不及MCF7-WT。该结果表明,FOXQ1 介导了缺氧引起的MCF7 细胞迁移能力的增加(图4)。
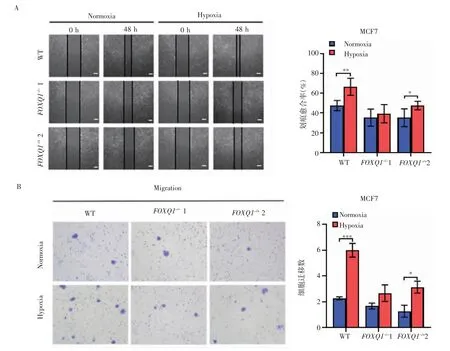
图4 FOXQ1 介导缺氧引起的MCF7 细胞迁移能力的增加Fig 4 FOXQ1 mediates the increased migration of MCF7 cells induced by hypoxia
2.5 HIF-1α 可能直接转录调控FOXQ1 的表达 分析来源于METABRIC 数据库的人类乳腺癌数据,发现FOXQ1 与HIF1A(r=0.099,P<0.000 1)以及HIF-1α 的代表性下游靶基因LOX[19](r=0.375,P<0.000 1)、LDHA[20](r=0.136,P<0.000 1)和GLUT3[21](r=0.213,P<0.000 1)的mRNA 表达有显著相关性;此外,在FOXQ1 的启动子上存在HIF-1α 的结合位点(图5)。

图5 HIF-1α 可能直接转录调控FOXQ1 的表达Fig 5 HIF-1α maydirectly regulate the expression of FOXQ1
3 讨论
自2013 年首次被应用于哺乳动物基因组编辑以来,CRISPR/Cas9 技术凭借其高效、精准的特点,被广泛应用于探索癌症相关基因的功能、建立荷瘤动物模型、探索药物靶点等,极大地增进了我们对癌症基因组学的认识[22-23]。在本研究中,笔者实践了一种更为方便、省时、高效的CRISPR/Cas9 解决方案,使用商品化Cas9 慢病毒和纯化的crRNA、tracrRNA 来构建CRISPR/Cas9 基因编辑体系,省去前期构建gRNA 载体或Cas9 载体的工作,极大的简化了基因编辑实验步骤。最终通过该技术,成功的构建了FOXQ1 基因敲除的MCF7 细胞株,克服了FOXQ1 在MCF7 中无法以siRNA 或shRNA 进行降表达的难题,并以此为模型在体外初步探究了FOXQ1 对缺氧环境中乳腺癌细胞迁移能力的影响。结果表明,缺氧能显著上调MCF7 细胞中FOXQ1的表达,同时也能在体外增加MCF7 细胞的迁移能力,并且该过程被FOXQ1 所介导。这初步印证了笔者的假设。
HIF-1α 是介导缺氧反应的中心转录因子,其已被广泛认为在肿瘤侵袭、转移中起关键作用[24]。在对临床数据的研究中发现,在乳腺癌中FOXQ1 与HIF1A 的mRNA 表达具有显著的相关性。同时,笔者还进一步分析了HIF-1α 的已知靶基因LOX、LDHA 和GLUT3 与FOXQ1 表达的相关性,发现仍然显著相关。并且FOXQ1 的启动子上存在HIF-1α的结合位点,因此推测FOXQ1 可能作为HIF-1α 的直接靶基因而被调节。
研究发现,Twist1、Snail、Slug、ZEB1 和ZEB2 等EMT 调节因子直接或间接受HIF-1α 的调控[25]。而据报道,Twist1、Snail 和ZEB2 也同样作为FOXQ1的下游因子调控肿瘤细胞的迁移侵袭[26-28]。同时,笔者的结果表明,FOXQ1 完全敲除后缺氧对MCF7 迁移能力的促进作用几乎完全被抑制,并且MCF7 细胞的迁移能力与FOXQ1 的表达具有一定的剂量依赖性,FOXQ1 在缺氧信号所调控的一系列细胞迁移相关下游因子中起关键作用。
总之,通过CRISPR/Cas9 系统成功构建了FOXQ1基因稳定敲除的乳腺癌MCF7 细胞株,并发现FOXQ1 介导了缺氧对MCF7 细胞迁移能力的促进作用。这些工作为今后深入研究乳腺癌的缺氧微环境中FOXQ1 的功能及其机制提供了良好的实验基础。
猜你喜欢
核科学与工程(2022年3期)2022-10-18
中国现代医药杂志(2020年10期)2020-12-14
广州化工(2020年6期)2020-04-18
现代矿业(2018年9期)2018-10-16
现代检验医学杂志(2016年3期)2016-11-15
医学研究杂志(2015年11期)2015-06-10
医学研究杂志(2015年3期)2015-06-10
特产研究(2015年1期)2015-04-12
中国当代医药(2015年16期)2015-03-01
中国医药导报(2015年27期)2015-02-28
