
Ultrasensitive proteomics depicted an in-depth landscape for the very early stage of mouse maternal-to-zygotic transition☆
2023-09-26 01:03LeiGuXumioLiWenchengZhuYiShenQinqinWngWenjunLiuJunfengZhngHuipingZhngJingqunLiZiyiLiZhenLiuChenLiHuiWng
Lei Gu , Xumio Li , Wencheng Zhu , Yi Shen , Qinqin Wng , Wenjun Liu ,Junfeng Zhng , Huiping Zhng , Jingqun Li , Ziyi Li ,*, Zhen Liu , Chen Li ,***,Hui Wng ,****
a Center for Single-Cell Omics, School of Public Health, Shanghai Jiao Tong University School of Medicine, Shanghai, 200025, China
b Institute of Neuroscience, CAS Center for Excellence in Brain Science and Intelligence Technology, CAS Key Laboratory of Primate Neurobiology, State Key Laboratory of Neuroscience, Chinese Academy of Sciences, Shanghai, 200031, China
c Shanghai Center for Brain Science and Brain-Inspired Intelligence Technology, Shanghai, 200031, China
d Shanghai Applied Protein Technology Co., Ltd., Shanghai, 201100, China
e University of Chinese Academy of Sciences, Beijing,101408, China
Keywords:
Single-cell proteomics
Low-input proteomics
Maternal-to-zygotic transition
Oocyte
Embryo
A B S T R A C T
Single-cell or low-input multi-omics techniques have revolutionized the study of pre-implantation embryo development. However, the single-cell or low-input proteomic research in this field is relatively underdeveloped because of the higher threshold of the starting material for mammalian embryo samples and the lack of hypersensitive proteome technology. In this study, a comprehensive solution of ultrasensitive proteome technology (CS-UPT) was developed for single-cell or low-input mouse oocyte/embryo samples. The deep coverage and high-throughput routes significantly reduced the starting material and were selected by investigators based on their demands. Using the deep coverage route, we provided the first large-scale snapshot of the very early stage of mouse maternal-to-zygotic transition,including almost 5,500 protein groups from 20 mouse oocytes or zygotes for each sample. Moreover,significant protein regulatory networks centered on transcription factors and kinases between the MII oocyte and 1-cell embryo provided rich insights into minor zygotic genome activation.
1. Introduction
Studies on oocyte meiotic maturation and pre-implantation embryonic development would provide informative and unique biological insights into mammalian reproduction and development.Oocyte meiotic maturation involves the resumption of meiosis and the completion of the first meiosis in primary oocytes arrested in the prophase of meiosis I or the germinal vesicle (GV) stage,followed by the initiation of the second meiotic division and arrest in metaphase II(MII)[1,2].After fertilization,the embryo undergoes a series of cleavage divisions during pre-implantation development, encompassing the 1-cell, 2-cell, 4-cell, 8-cell, morula, and blastocyst stages [3]. This developmental process is a complex biological stage involving critical events, such as zygotic genome activation (ZGA) and first cell lineage differentiation [3,4]. Fertilization triggers a remarkable,complex cell fate transition in oocytes from terminal differentiation to the totipotent state, termed the maternal-to-zygotic transition (MZT). MZT is a conserved, fundamental process during which the maternal environment of oocytes transitions to a zygotic genome-driven expression program, and terminally differentiated oocytes and sperm are reprogrammed to totipotency.During this period,it is crucial to fine-regulate events,e.g., maternal and zygotic transcript storage, timely decay, and sequential translation activation [5-10]. Several studies based on single-cell or low-input multi-omics technologies have deciphered this complex and mysterious developmental process at the genetic[11-14], epigenetic [15,16], translational [4,11,17], and metabolic[15,18] levels to obtain valuable and full-scale biological insights.
MII oocytes and zygotes serve as mature oocytes and initiate pre-implantation embryo development, respectively, and characterizing their molecular landscapes at different levels plays a critical role in uncovering MZT and ZGA in mammals.Minor ZGA must precede major ZGA to execute the MZT successfully,and the timely occurrence of minor ZGA is crucial for pre-implantation development to continue beyond the two-cell stage [19]. Studies, such as those conducted by Asami et al. [13], have revealed that human embryonic transcription initiates at the 1-cell stage via the singlecell RNA sequencing of human MII oocytes and 1-cell embryos.Zhang et al. [4] showed that mouse MII oocytes and zygotes were similar at the RNA level but differed markedly at the translational level. Transcription factors (TFs) are critical for MZT [5,7]. Xiong et al. [17] found that some transcriptional regulators were translationally repressed in mouse MII oocytes and re-upregulated in 1-cell embryos. Our recent work also demonstrated that maternally inherited mRNAs with m6A modifications are sustained throughout the MZT and contribute to protein synthesis,which may be crucial for MZT and pre-implantation development in mice [20]. Nevertheless, proteins are the actual executors of most biological processes, and gene expression during development requires confirmation at the protein level [3]. A growing body of literature has recognized the poor correlation between transcriptomic and proteomic data at the same developmental stage [21-23]. Therefore, delineating the proteomic landscape involved in this developmental process is necessary.
Mass spectrometry (MS) is a powerful tool for comprehensive and unbiased proteomic analysis [24,25]. Several studies have explored the proteome of oocyte meiotic maturation and preimplantation embryo development [26-34]. For example, in 2010, Wang et al. [27] collected 7,000 mouse oocytes at different developmental stages and identified 2,781, 2,973, and 2,082 proteins in the GV oocytes, MII oocytes, and fertilized oocytes (zygotes), respectively. Six years later, another study successfully reduced the starting material to 600 mouse embryos and identified an average of more than 5,000 proteins[35].With the development of technology, the threshold for mouse embryo proteomics research has been greatly reduced,and the accessibility of reaching the number of mouse embryos required for a single replicate has increased.However,a biologically meaningful experimental design may require using a relatively large number of embryos because of problems such as experimental repeatability and control condition settings. In addition, the additional starting materials increase the cost of the experimental process. Therefore, the relatively high experimental cost has discouraged many researchers and limited the development of related fields. However, with the progress of MS and the innovation of single-cell analysis strategies,illustrating the protein profiling of trace or single cells based on MS has become possible. From one mammalian somatic cell, more than 2,000 proteins can be reliably quantified [36-38]. Several studies have applied single-cell proteomic techniques to oocyte maturation.Guo et al.[23]quantified approximately 1,900 proteins on average from one human oocyte. However, the size of oocytes differs between mice and humans, resulting in an approximately four-fold difference in protein content[39],increasing the difficulty of illustrating the proteomic landscapes of single mouse oocytes. Using an ingenious single-cell proteomic strategy, a nanoliter-scale oil-airdroplet (OAD) chip, Li et al. [40] identified 355 proteins from a single mouse oocyte.Using the same preparation methods as those aforementioned for human oocytes, Guo et al. [23] identified no more than 500 protein groups in a single mouse oocyte. These proteomic depths are far from the starting point for solving biological problems. It further highlights the scarcity and importance of biomaterials derived in vivo and the need to develop new methods to completely characterize single-cell, low-input oocyte,or embryo proteomes.
In this study, we report a comprehensive solution of ultrasensitive proteome technology (CS-UPT) for single-cell or low-input mouse embryo samples (Fig. 1). Both deep coverage and highthroughput routes were able to significantly reduce the starting material and be selected by investigators based on their demands.In brief, our ultrasensitive proteomic platform quantified an average of 2,665 protein groups in a single zygote. When the starting material was increased to 20 zygotes,the average number of quantified proteins increased to 4,585. To our knowledge, the early stages of mouse MZT lack a large-scale proteomic landscape using a low-input approach. Thus, using our deep coverage technique,we obtained a proteomic snapshot of mouse MII oocytes and zygotes.As a result,5,455 protein groups were identified,of which 91% (4,977 protein groups) provided reliable quantitative data.Notably, significantly regulated 33 TFs, 37 kinases, and two subcortical maternal complex (SCMC) members were found, indicating that their protein-protein regulatory network played critical roles at the very early stage of MZT.

Fig. 1. A comprehensive solution of ultrasensitive proteome technology (CS-UPT) for single-cell or low-input mouse embryo samples. The top is the deep coverage strategy,prepared in tubes without labeling.The bottom is the high-throughput strategy,prepared in 384-well plates with tandem mass tags(TMT)labeling.ABC:ammonium bicarbonate;LC-MS/MS: liquid chromatography tandem mass spectrometry.
2. Materials and methods
2.1. Reagents and chemicals
Pregnant mare serum gonadotropin (PMSG) and human chorionic gonadotropin (hCG) were obtained from San-Sheng Pharmaceutical Co.,Ltd.(Shenyang,China).4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), glycerol, Triton X-100, Tween 20,NP-40, deoxycholate, ethylene diamine tetraacetic acid (EDTA),NaCl,tris(2-chloroethyl)phosphate(TCEP),chloroacetamide(CAA),ammonium bicarbonate (ABC), trifluoroacetic acid (TFA), K+Simplex Optimised Medium (KSOM), M2 medium, hyaluronidase, and acidic Tyrode's solution were obtained from Sigma-Aldrich (St.Louis, MO, USA). Sodium dodecyl sulfate (SDS), formate, acetonitrile (ACN), magnetic beads, MS-grade trypsin, TMTsixplex™reagents,and TMTpro™16plex reagent were obtained from Thermo Fisher Scientific Inc. (Waltham, MA, USA). Phosphate buffer saline(PBS) was obtained from Solarbio (Beijing, China). Ethanol (EtOH)was obtained from Sinopharm Chemical Reagent (Beijing, China).
2.2. Starting materials
2.2.1. Mouse
All animal experiments involving mice were performed in accordance with the guidelines of the Animal Care and Use Committee of the Institute of Neuroscience, Center for Excellence in Brain Science and Intelligence Technology, Chinese Academy of Sciences,Shanghai,China(Formal approval No.:NA-042-2019 and NA-042-2022). The mice were maintained in Assessment and Accreditation of Laboratory Animal Care, an accredited specific pathogen-free facility, under a 12 h light, 12 h dark cycle. The ambient temperature was 20°C,and the relative humidity was 50%.
2.2.2. Oocyte and zygote collection
Mouse MII oocytes were derived fromwild-type B6D2F1(C57BL/6 J×DBA/2 N)strain female mice[41].For superovulation,8-weekold female mice were injected intraperitoneally with 10 IU PMSG,followed by the injection of 10 IU hCG 48 h later.MII oocytes were collected from the oviducts of the superovulated female mice from 14 to 16 h after hCG injection,and cumulus cells were removed from the oocytes by brief incubation in M2 medium with hyaluronidase.For mouse oocytes without the zona pellucida (ZP), the ZP was removed by washing the MII oocytes with acidic Tyrode's solution using a mouth pipette.After the disappearance of the ZP,the oocytes were washed with M2 medium and PBS for further tests.
For zygote collection, superovulated B6D2F1 female mice were mated with adult B6D2F1 males, and zygotes were collected from female oviducts from 14 to 16 h post-hCG injection. Zygotes with two pronuclei were transferred from M2 medium to KSOM and cultured at 37°C under 5% CO2. One-cell stage embryos were collected from 27 to 29 h after hCG injection.
2.3. Sample preparation in tube for data-independent acquisition(DIA) and data-dependent acquisition (DDA) analysis
Clean zygotes or oocytes were collected in tubes containing lysis buffer(50 mM HEPES pH=8,1%SDS,1%glycerol,1%Triton X-100,1%Tween 20,1%NP-40,1%deoxycholate,5 mM EDTA,and 50 mM NaCl)and lysed at 60°C for 2 h[42].TCEP and CAA were added at final concentrations of 10 and 40 mM, respectively, and incubated for 5 min at 95°C to reduce and alkylate proteins. Prewashed magnetic beads were added to the protein solution at a bead/protein ratio of approximately 10:1 (m/m). EtOH was added to a final concentration of 50%, and the protein-beads mixtures were incubated at 1,200 r/min at room temperature(RT)to promote protein binding. The supernatant was removed on the magnetic rack as a waste.The protein-binding beads were washed twice with 80%(V/V) EtOH, followed by another wash with 100% EtOH, to remove detergents and other contaminants. 200 ng of MS-grade trypsin dissolved in 100 mM ABC was added to resuspend the proteinbinding beads. After enzymolysis at 1,200 r/min at 37°C for 4 h,the supernatant containing digested peptides was transferred to clean tubes and acidized. The samples were then desalted using homemade C18StageTips[43].Clean peptides were lyophilized and resuspended in 0.1% (V/V) formate/ddH2O for liquid chromatography tandem-MS (LC-MS/MS) analysis.
2.4. Sample preparation in 384-well plate for tandem mass tag(TMT)-label analysis
Single zygotes were dispensed in 384-well plates (Eppendorf,Hamburg, Germany) containing 0.1% (V/V) RapiGest. The plates were frozen at -80°C overnight and then heated to 90°C for 10 min to lyse the cells and release the proteins by freeze-thawing.MS-grade trypsin in HEPES was added at a final concentration of 10 ng/μL,and the 384-well plates were incubated at 37°C for 4 h to digest the proteins.Peptides were then labeled with TMTsixplex™reagents or TMTpro™16plex reagent in 40%(V/V)ACN for 1 h at RT and quenched by 0.2% (V/V) TFA. Differentially labeled samples from one TMT set were combined and desalted using homemade C18StageTips.Clean peptides were lyophilized and resuspended in 0.1% (V/V) formate/ddH2O for LC-MS/MS analysis.
For the TMT6plex set,single zygotes were labeled with 126,127,128,and129 channels.The carrier samplewas prepared in bulk using a filter assisted sample preparation (FASP) procedure [44], which used 1,000 oocytes as the starting material.Peptides equivalent to 50 or 100 oocytes were dissolved in 0.1%(V/V) RapiGest and prepared using the same steps as those for single-cell samples synchronously and labeled with the TMT 131 channel.Blank samples were prepared using the same steps as those for the single-cell samples synchronously without any cell or peptide addition as the starting material and were labeled with the TMT 130 channel.For the TMT8plex set,single zygotes were labeled with eight channels from 16plex of TMTpro™:126,127N,128C,129N,130C,131N,132C,and 133N.
2.5. LC-MS/MS analysis
All samples were separated via a high performance applied chromatographic system nanoElute®(Bruker Daltonics, Bremen,Germany). Peptides were loaded onto an in-house packed column(75 μm × 250 mm; 1.9 μm ReproSil-Pur C18beads, Dr. Maisch GmbH,Ammerbuch,Germany)heated to 60°C and separated with a 60-min gradient from 2%to 80%mobile phase B at a flow rate of 300 nL/min. The mobile phases A and B were 0.1% (V/V) formate/ddH2O and 0.1% (V/V) formate/acetonitrile, respectively.
MS analysis was performed using a trapped ion mobility spectrometer (TIMS) coupled to a time-of-flight mass spectrometer(timsTOF MS, Bruker Daltonics). Parallel accumulation-serial fragmentation(PASEF)was applied in all sample analyses[45].MS and MS/MS spectra were recorded from m/z 100 to 1,700, and the quadrupole isolation width was set to 2 Thomsom (Th) for m/z < 700 and 3 Th for m/z > 700. For TMT-labeled and label-free samples acquired in the DDA mode, the duration of one topN acquisition cycle was 100 ms and contained one MS1 survey TIMSMS and eight PASEF MS/MS scans with two TIMS steps.
For label-free samples acquired via DIA, we set four precursor isolation window schemes for fragment analysis. Method 1 contained 64 × 26 Th precursor isolation windows from m/z 400 to 1,200 with an isolation width of 1 Th, of which one mass-width window contained two mobility windows and one MS1 scan,followed by 16 consecutive MS2 scans.Method 2 contained 36×27 Th precursor isolation windows from m/z 300 to 1,200 with a 2 Th isolation width overlap,of which one MS1 scan was followed by 18 consecutive MS2 scans. Method 3 contained 39 × 22 Th precursor isolation windows from m/z 400 to 1,180 with a 2 Th isolation width overlap,of which one MS1 scan was followed by 13 consecutive MS2 scans. Method 4 contained 16 × 52.5 Th precursor isolation windows from m/z 400 to 1,200 with 2.5 Th isolation width overlap,of which one MS1 scan was followed by four consecutive MS2 scans and every MS2 scan was repeated four times. Details of the DIA parameters are listed in Table S1.In all scan modes,collision energy was ramped linearly as a function of the mobility from 59 eV at 1/K0=1.6 Vs/cm2to 20 eV at 1/K0=0.6 Vs/cm2.
2.6. Database searching
MS raw files of TMT-label experiments and DDA label-free experiments were processed with SpectroMine™ (V3.2; Biognosys AG, Schlieren, Switzerland) and searched against the UniProt/SwissProt mouse database (UP000000589, containing 17,090 entries, downloaded on Nov 24, 2021) using the Pulsar engine.For DDA analysis,the default factory settings were used.In brief,trypsin/P was set as the digestion enzyme,with a maximum of two mis-cleavage sites allowed. False discovery rates were controlled at 1%for the peptide and protein groups.Only peptides with lengths ranging from 7 to 52 amino acids were considered for the search.The oxidation of methionine and the acetylation of the protein N-term were set as variable modifications, and carbamidomethyl of cysteine was set as a fixed modification. For TMT6plex-label analysis, similar default settings were used,except for the calibration MS1 and MS2 mass tolerances, which were set to 20 ppm. For TMT8plex-label analysis, quantification was based only on the reporter ions of eight label channels (126,127N, 128C, 129N, 130C, 131N, 132C, and 133N) from TMTpro™,and the calibration MS1 and MS2 mass tolerances were set to dynamic as the default.
DIA label-free data were processed with Spectronaut®(V16;Biognosys AG). For the directDIA search, raw files were searched against the UniProt/SwissProt mouse database as same as DDA analysis with default parameters. A hybrid spectral library of mouse oocytes and the pre-implantation embryo proteome was established using 10 DDA runs and 41 DIA runs containing 140,511 precursors, 96,036 peptides, and 6,632 protein groups.For the library-based DIA search, raw files were searched against this self-established hybrid spectral library by using the default parameters.
MS proteomics data have been deposited to the ProteomeXchange Consortium [46] via the PRoteomics IDEntification database (PRIDE) [47] partner repository with the dataset identifiers PXD041399 and PXD041023. For PXD041023, we researched six related raw datasets(MII_1-3 and 1-cell_1-3)and analyzed the proteomic results in detail. The proteome from MII_1, MII_2,1 cell_1,1 cell_2, late2_2, and late2_3 in PXD041023 was used for the analysis and was recently published as a partial dataset in our other work[20].
2.7. Data analysis
Statistical analyses and visualizations were performed using R(V4.2.2) unless otherwise stated. Data are presented as mean value ± standard deviation. The distribution of the parallel accumulation-serial fragmentation combined with data-independent acquisition (diaPASEF) isolation windows was visualized using DataAnalysis software (V5.3, Bruker Daltonics). The Venn diagram was generated by jvenn [48]. In the comparative ZP experiments and MZT experiments, protein groups with quantitative information in at least two replicates from each group were defined as reliable quantitative datasets for further bioinformatics analysis.Missing data values were not imputed.After median normalization and Log2 transformation, reliable quantitative data were statistically analyzed by Student's t-test or the Wilcoxon test according to whether the data fit the normal distribution. Differentially expressed proteins(DEPs)were sorted using a fold change cutoff of 1.2 and a P value cutoff of P < 0.05. For stage-specific proteins analysis in MII oocytes and 1-cell embryos, protein groups that were not quantified across all replicates in one group but were quantified in at least two replicates in the other group were defined as stage-specific proteins.A hypergeometric test(phyper)in R was used to assess the significance of different DEP groups. Functional enrichment analysis was performed for gene ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG)pathway analysis via the Metascape (http://www.metascape.org/)online tool [49]. Analyses were conducted with the default enrichment parameters,among which the minimum overlap was 3,P value cutoff was 0.01, and the minimum enrichment was 1.5.Kinase annotation was performed using Metascape online tool. As for TFs annotation, both the Mouse Integrated Protein-Protein Interaction rEference (MIPPIE, https://cbdm.uni-mainz.de/mippie)and the Transcriptional Regulatory Relationships Unraveled by Sentence-based Text mining (TRRUST, https://www.grnpedia.org/trrust/) databases were used [50,51]. Cytoscape (V3.9.1, http://www.cytoscape.org/) [52] was used to determine the network characteristics of DEPs based on MIPPIE, TRRUST, and Search Tool for the Retrieval of Interaction Gene/Proteins (STRING, http://string-db.org/). Molecular complexes in the subnetwork dominated by kinases and TFs were selected by Molecular COmplex DEtection (MCODE, V2.0.2) in Cytoscape using the default parameters,then annotated by Metascape.
3. Results
3.1. Establishing optimal diaPASEF acquisition scheme to depict zygote proteome
Optimized diaPASEF acquisition schemes with different emphases can achieve a balance among selectivity, sensitivity, and precursor coverage [53]. Properly setting precursor isolation windows can increase the data acquisition efficiency and maximize ion utilization. To achieve high data integrity and deep proteomic coverage of mouse zygotes,we employed four diaPASEF acquisition schemes using timsTOF Pro (Table S1 and Fig. S1). Method 1 used the standard isolation window setting reported by Meier et al.[53]and contained 64 × 26 Th precursor isolation windows (Fig. S1A).Method 2 was automatically optimized using the analysis software DataAnalysis,based on the previous DDA data of the same sample,and contained 36 × 27 Th precursor isolation windows (Fig. S1B).Methods 3 and 4 were inspired by Meier et al. [53] and contained 39 × 22 Th and 16 × 52.5 Th precursor isolation windows,respectively (Figs. S1C and D). Applying these four diaPASEF acquisition schemes, we detected an average of 19,959, 17,156,19,642, and 14,096 peptide precursors from one mouse zygote in duplicate runs (Fig. S1E). The proteomic coverage depth of each scheme reached 2,375, 2,151, 2,313, and 1,966 protein groups on average (Fig. S1F). Overall, among the four methods, method 1 achieved the highest number of precursor identifications, the deepest proteomic coverage,and the lowest coefficient of variation(Fig. S1G). Therefore, we chose method 1 for subsequent protein profiling of mouse zygotes.
3.2. Low-input proteomic workflow ensuring deep coverage in mouse oocytes with zona pellucida
The ZP is a gel-like structure surrounded the oocytes and preimplantation embryos.The mouse ZP consists mainly of three proteins (ZP1, ZP2, and ZP3) [54-56]. However, these three proteins represent approximately 15%of all oocyte proteins[57,58].Although the dynamic range of MS is from four to six orders of magnitude,the signals of high-abundance proteins can largely suppress the signals of low-abundance proteins owing to the mask effect in complex samples [59]. Therefore, most published proteomic studies have removed the ZP from oocytes or pre-implantation embryos before cell lysis [30-35]. Notably, less researches have explored whether the removal of the ZP would affect the oocyte proteome.
Thus,we compared the proteomic landscape of oocytes with and without the ZP and explored whether our workflow could acquire deep coverage proteomic information from ZP-intact oocytes.After treatment with acidic Tyrode's solution,ZP-free MII oocytes(NoZP group)were prepared and analyzed using the same workflowas that for the ZP-intact MII oocytes(ZP group)(Fig.S2A).The sum intensity of ZP proteins(ZP1,ZP2,and ZP3)identified from ZP-intact oocytes made up an average of 4.26%of the total protein intensity,whereas the NoZP group could hardly detect any ZP proteins (Fig. S2B). A similar number of protein groups were identified in oocytes with or without the ZP(Fig.S2C),and most proteins(99.84%)were identified in both groups(Fig.S2D).These results convincingly show that the high-abundance proteins in the ZP did not decrease the detection depth with our workflow. Notably, the intensities of ZP1, ZP2,and ZP3 represented approximately 4.1%,49.8%,and 46.1%of total ZP proteins, respectively (Fig. S2E). This proportion was similar to published results [54,56], which confirmed the quantitative accuracy of our data.However,the coefficient of variation of NoZP group was higher than that of the ZP group(Fig.S2F),demonstrating that the additional step of ZP removal may introduce non-negligible interference caused by manual operation, especially in trace biological systems. From the 3,015 proteins identified in at least two samples from each group,151 DEPs were found between the two groups(P<0.05,fold change ≥1.2),of which 76 were upregulated in the ZP group,and 75 were upregulated in the NoZP group(Fig.S2G).Unsurprisingly, compared with ZP-free oocytes, ZP3, ZP2, and ZP1 were the top three upregulated proteins in ZP-intact oocytes.Mfge8(lactadherin) and Apoa1 (apolipoprotein A-I) were highly upregulated in the ZP group(Fig.S2G).Mfge8 is a ZP-binding protein that mediates sperm-egg binding [60]. Apoa1 is a component of the sperm-activating protein complex(SPAP)and may play a role in the regulation of metabolic pathways during blastocyst formation and cell differentiation[61,62].The upregulated proteins in each group were enriched in different biological processes (Fig. S2H). In brief,the elevated proteins in ZP-free oocytes were enriched in metabolic and biosynthesis-related pathways,whereas those in ZP-intact oocytes were enriched in lipid storage, actin cytoskeleton organization,sperm-egg recognition,and fertilization.Thus,we inferred that removing the ZP might influence some proteins and pathways related to reproduction and development in MII oocytes. In summary, because our low-input proteomic workflow has a similar performance in oocytes with or without the ZP and that ZP removal may introduce some interference to the oocyte proteome,we chose oocytes or embryos with the ZP for subsequent studies.
3.3. Single-cell and low-input proteomics deepens zygote proteomic coverage while significantly reducing starting material
Using the aforementioned optimal diaPASEF acquisition scheme,we performed DIA-based proteomic analysis of 1, 2, 5, 10, and 20 ZP-intact mouse zygotes. A total of 4,700 protein groups were identified, of which an average of 2,665, 3,291, 3,893, 4,223, and 4,585 protein groups were detected in two replicates of 1, 2, 5,10,and 20 zygotes,respectively(Fig.2A).Remarkably,even in a single mouse zygote,an average of 2,665 protein groups was be identified.We found that the correlation between the starting number of zygotes and the number of identified protein groups could be well fitted as a logarithmic curve(R2=0.9848),which performed better than a linear fitting(R2=0.7741)(Fig.2A).This result indicates that although the number of identified protein groups increased with increasing starting materials, the benefit progressively decreased.When the starting material was increased to 30 mouse zygotes,the peptide levels exceeded the recommended optimal detection range(≤200 ng)for timsTOF Pro(manufacturer's recommendations),and the number of protein groups identified increased very limited(data not shown). Notably, compared with searching by directDIA,protein identification can benefit from a project-specific spectral library, especially as the amount of starting material increases(Fig. S3A). The proportion of co-identified proteins in the two replicates of each group to the total identified proteins and the Pearson's correlation coefficients between 10 runs of zygotes revealed the excellent repeatability and stability of the proteomic data (Figs. 2B and C). Notably, when the starting material was increased from one to 20 zygotes, the proportion of proteins that could be repeatedly detected in two replicates increased from 77.8%to 96.3% (Fig. 2B). The proteomic data showed good repeatability even for one zygote (Fig. 2B). However, as starting material increases, the repeatability of proteomic data increases, which also suggests that the tinier the experimental system, the greater the likelihood that interferences (e.g., instrumental, or manually operated factors) would disturb the results.

Fig.2. Data-independent acquisition(DIA)-based low-input proteomics improves zygote(Zyg)proteome depth while significantly reducing starting material.(A)Number of protein groups identified from mouse zygotes of various numbers.Each group had two independent replicates(10 runs);data in the plot are presented as mean±standard deviation(SD).Curve Fitting was performed with R(V4.2.2).(B)Proportion of co-identified proteins between the two replicates in each group.(C)Correlation of protein expression among 10 mass spectrometry (MS) runs of mouse zygotes. There were five groups with different numbers of zygotes and two replicates in each group. Pearson's correlation coefficients were presented numerically and color-coded. (D) Comparison of the number of commonly identified proteins and uniquely identified proteins between different numbers of zygotes.Unique proteins were defined as being only identified in one group and not identified in all other groups. (E) Intensity distribution of 471 unique proteins identified in 20 mouse zygotes. (F) Intensity distribution of 2,273 proteins co-identified in five groups with different zygote numbers. (G) Doughnut chart displays the distribution of top 10 subcellular compartments for identified proteins in each group based on enrichment analysis of Gene Ontology (GO) cellular component terms.
Next, we explored the overlap of protein groups in different starting zygote numbers and found that the 20 mouse zygotes identified 471 unique proteins, which were predominantly lowabundance proteins and mainly participated in mitochondrial translation and RNA metabolic processes (Figs. 2D, 2E, S3B, and S3C). A total of 2,273 proteins were commonly identified in all samples, covering approximately the top 80% of the protein intensities (Fig. 2F). In addition, even with a single zygote starting material, protein expression remained in a wide intensity range,but adding starting materials increased the integrity of the proteomic data (Figs. S3D-H). Based on the enrichment analysis of the GO cellular component terms, the subcellular distributions of proteins identified in different numbers of zygotes were very similar, illustrating that our characterization of the zygote proteome was unbiased (Figs. 2G and S3I-K).
We also tested the same proteomic preparation workflow using DDA-mode LC-MS/MS analysis, achieving an average of 1,972 and 3,437 identified protein groups from one and 20 mouse zygotes,respectively (Fig. S4A). Although the protein identification performance and data correlation were not as satisfactory as those in DIA mode(Fig.S4),these results provided outstanding proteome depth and can assist in building project-specific spectral libraries.
In summary,even when only one zygote was used as the starting material, the protein profile was reliable and sufficiently deep for downstream analysis. However, if a study requires more focus on the depth of proteomic data and repeatability of quantification,more starting material are recommended. In short, this requires balancing data integrity and quantitative repeatability with the starting material consumption.
3.4. High-throughput protein profiling of mouse zygotes by multiplexed single-cell proteomics
Isobaric label-based quantification is one of the most used proteomic quantification strategies and has been widely applied to improve analysis throughput and quantitative accuracy [63-68].TMT-labels are among the most widely used isobaric labels in proteomics and have been expanded to several serials, including 6plex, 10plex, 11plex, 16plex, and 18plex [69-71]. Slavov and coworkers [72] pioneered the application of TMT to single-cell proteomics and introduced the idea of a “carrier channel”, which consisted of approximately 200 cells to share most of the loss from single-cell channels and enhance the signal for MS analysis.Several single-cell proteomic tools have been developed based on this labeling strategy, including the ultra-sensitive and easy-to-use multiplexed single-cell proteomic workflow (UE-SCP) built in our lab [37]. Because of its excellent performance in mammalian somatic cells, we attempted to apply a similar strategy to the single mouse zygote proteomics. First, we designed a set of 6-channel experiments based on TMT6plex (Fig. 3A). Each TMT set included four channels containing one zygote in each channel (single-cell channel), one channel without any cells or peptides (blank channel),and one carrier channel consisting of approximately 50 or 100 mouse oocytes(50×carrier or 100×carrier channel)(Fig.3B).The 50× carrier experiment and 100× carrier experiment were repeated twice,and an average of 2,182 or 2,371 protein groups was identified from one zygote in the 50×or 100× carrier experiment group, respectively (Fig. 3C). A boxplot of protein intensities in different channels showed good data parallelism between the single-cell channels, and the low intensity of the blank channel confirmed that the influence of the carrier channel was minimized(Fig. 3D). The stability of the defined quantitative ratios of singlecell channels illustrated that in the 50× carrier or 100× carrier experiment, the quantification of single-cell channels was reliable(Fig. 3E). Although the number of proteins identified from one mouse zygote was not as high as in the aforementioned DIA-based label-free method, the label-based data showed a higher correlation between different single zygotes (Fig. 3F), which is a wellknown benefit of label-based proteomics [73]. More importantly,multiplexed labeling increased quadruple throughput of the MS analysis. Meanwhile, transferring the preparation processes from tubes to 384-well plates can excitedly enhance efficiency by more than 300-fold (Table 1). To obtain 300 single zygote proteomic datasets, the DIA-based method requires 25.42 days with high labor intensity, but the TMT6plex label-based method only requires 8.17 days. This throughput makes large-scale proteomics apply in fields of oocyte maturation and pre-implantation embryo development possible.
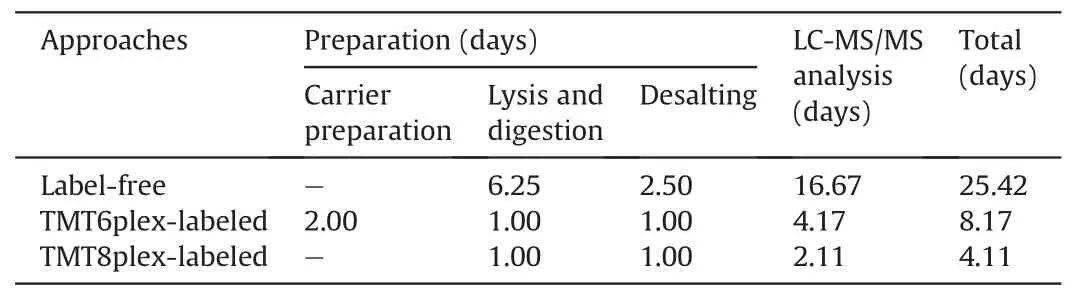
Table 1 The estimated time for three proteomic approaches to analysis 300 single mouse zygotes.
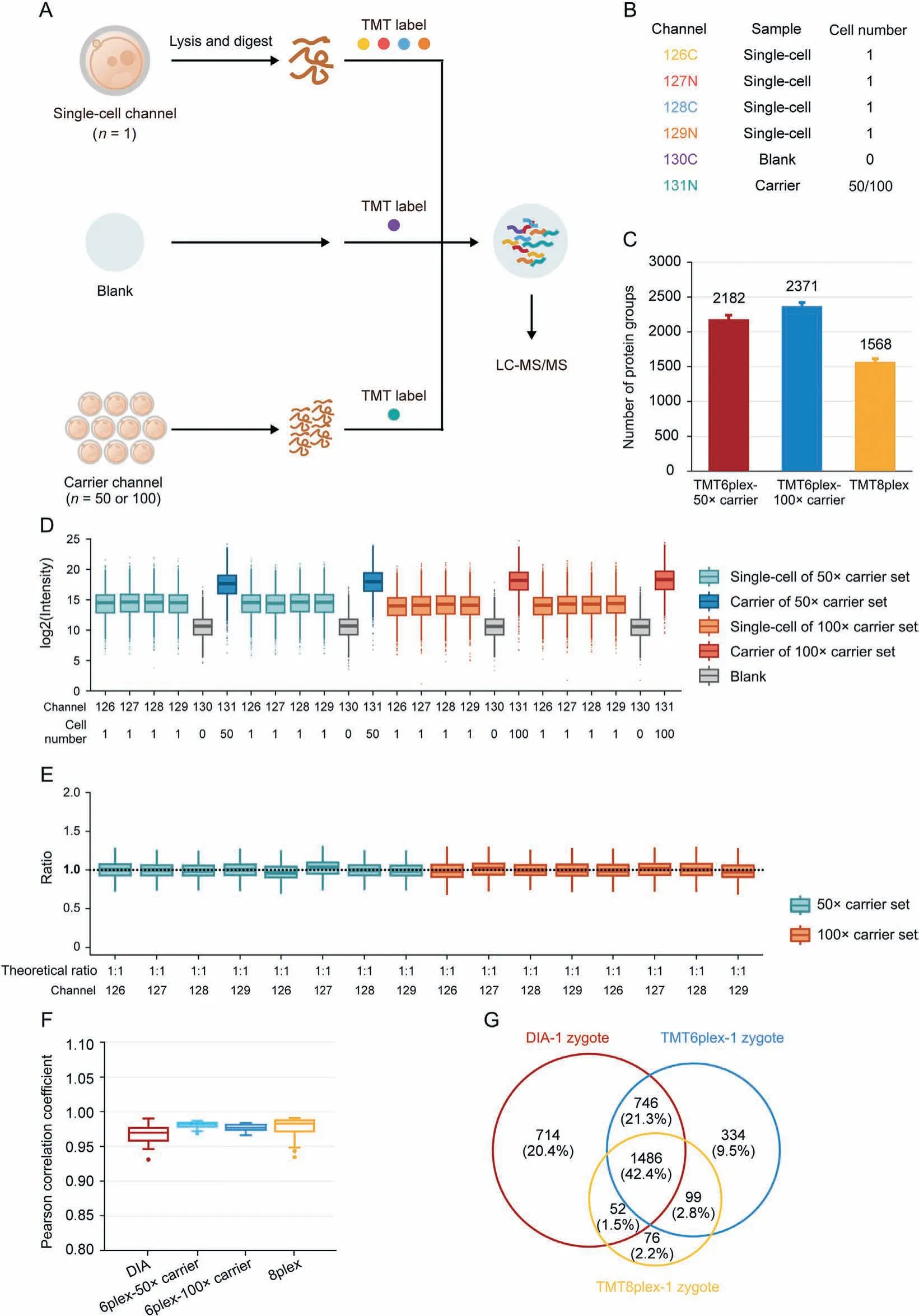
Fig.3. High-throughput proteomic profiling of mouse zygotes by single-cell proteomics.(A)Workflow for characterizing the mouse zygote proteome by high-throughput single-cell proteomics with the carrier channel.(B)Channel design for TMT6plex single-cell proteomics.Each TMT6plex experiment included one blank channel(0 zygote),one carrier channel(50 or 100 oocytes), and four single-cell channels (1 zygote for each). (C) Comparison of the number of protein groups identified by multiplexed single-cell proteomics with TMT6plex-50×carrier, TMT6plex-100×carrier, and TMT8plex. Data in the plot are presented as mean ± standard deviation. (D) Intensity distribution of all proteins in each TMT channel. Each different carrier TMT6plex experiment had two replicates. Boxplots show the median (middle bar), 25th and 75th percentiles (box), and 1.5× interquartile range(whiskers). (E) Boxplots of measuring ratios of protein groups based on tandem mass tags (TMT) intensities between different single-cell channels in each TMT6plex experiment.The denominator is the quantitative mean of 16 single-cell channels in four TMT experiments. Boxplots show the median (middle bar), 25th and 75th percentiles (box), and 1.5×interquartile range(whiskers).(F)Comparison of Pearson's correlation coefficients between each run(or single-cell channel)of data-independent acquisition(DIA)low-input proteomics, TMT6plex-50× carrier, TMT6plex-100× carrier, and TMT8plex single-cell proteomics. Boxplots show the median (middle bar), 25th and 75th percentiles (box), and 1.5× interquartile range (whiskers). (G) Venn diagram shows the overlap between DIA low-input proteomics, TMT6plex, and TMT8plex single-cell proteomics identified proteins from single mouse zygotes. LC-MS/MS: liquid chromatography tandem mass spectrometry.
Although carrier channels effectively improve protein identification in a single cell,their adverse impact on quantitative accuracy is still debatable [74]. Our data also confirmed that the more cells used in the carrier channel, the lower the correlation between single-cell channels (Fig. 3F). The quantitative ratio between the carrier and single-cell channels revealed a compressibility effect caused by the large intensity difference among the different TMT reporter ions(Fig.S5A).Thus,we attempted to eliminate the carrier channel and increase the number of channels to eight to enhance the total signal for MS analysis (Fig. S5B). An average of 1,568 protein groups were identified from a single mouse zygote in TMT8plex experiment without carrier (Fig. 3C), and the data parallelism was good (Fig. S5C). As expected, the correlations of single zygotes were better than those with TMT6plex experiment with 50× or 100× carrier (Fig. 3F). A total of 1,486 proteins were identified in a single mouse zygote using the DIA-,TMT6plex-,and TMT8plex-based methods (Fig. 3G), which validated the reliability of the data. It is worth mentioning is that using the TMT8plexbased strategy, analyzing 300 single zygotes required only approximately 4.11 days (Table 1), which provides a higher throughput option for researchers.
3.5. First large-scale snapshot for very early stage of mouse MZT by low-input proteomic approach
To further investigate whether our proteomic strategy would provide adequate information for real biological discovery, we treated mouse MII oocytes and zygotes(1-cell stage embryos)using a DIA-based low-input proteomic workflow to explore proteomic changes during the very early stage of mouse MZT. Each stage contained three replicates, with each replicate containing approximately 20 oocytes or 1-cell embryos.In total,5,455 protein groups were identified from six samples (Table S2), reaching a depth that usually required hundreds or thousands of oocytes or embryos[26-29,33-35].The average number of protein groups identified in each sample reached 5,115 (Fig. 4A). The standardized data also maintained a similar distribution, indicating high data quality(Fig.4B).Wang et al.[35]conducted a proteomic work that focused on MII oocytes and 1-cell embryos. Notably, 65.8% of proteins(2,786) identified from their data were observed in our work, and an additional 2,191 proteins were identified in our work, with the starting material of one replicate decreasing 30-fold (Fig. 4C). We found that the proteins identified in these two stages showed a high degree of consistency (96%, 5,231 protein groups), but some specific proteins were still expressed at each stage (Fig. 4D). The results of principal component analysis (PCA) also showed good intragroup repeatability and clear variations in the two stages(Fig.S6A),indicating that the proteome changed significantly after fertilization.
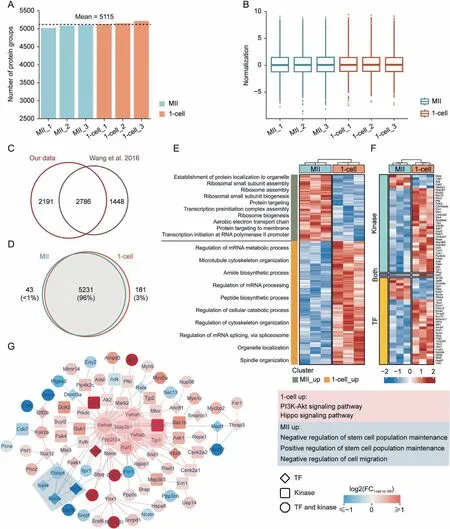
Fig. 4. Proteomics analysis of very early stage of mouse maternal-to-zygotic transition by ultrasensitive proteomics. (A) Protein groups identified in each sample. (B) Data distribution after normalization for each sample.(C)Comparison of protein groups identified in this experiment and Wang et al.[35].(D)Venn diagram of protein species in metaphase II (MII) oocytes and 1-cell embryos. (E) Heatmap and biological processes annotation of differentially expressed proteins. (F) Differentially expressed kinases and transcription factors(TFs)identified in this experiment.(G)Differentially expressed protein interaction network dominated by kinases and TFs containing major molecular complexes formed by their respective upregulated proteins;colored areas are molecular complexes detected by Molecular COmplex DEtection(MCODE);corresponding boxes are functional annotations.mRNA: messenger RNA; PI3K: phosphatidylinositol-4,5-bisphosphate 3-kinase; Akt: protein kinase B; FC: fold change.
Findings in the literature have suggested that some proteins appear de novo during early mouse embryogenesis [75,76]. Based on the proteomic data, we identified 28 stage-specific proteins in MII oocytes and 105 stage-specific proteins in 1-cell embryos(Table S2). Protein function analysis demonstrated that MII oocyte stage-specific proteins were mainly enriched in the ubiquitindependent protein catabolic process, the modification-dependent protein catabolic process, protein maturation and modification,which was consistent with findings in the literature that the ubiquitin-proteasome pathway modulated mammalian oocyte meiotic maturation and fertilization[77-79].Comparatively,1-cell embryo stage-specific proteins were mainly enriched in the developmental growth-, growth-, and regeneration-related pathways,suggesting that these proteins emerging de novo at the 1-cell stage are essential for early mouse embryonic development(Figs. S6B and C).
4月11日,北京市商务委员会发布《关于申报2018年度第一批商务发展项目的通知》,对商务发展领域内18个方向的项目进行资金补助。重点支持商务发展领域内促进生活性服务业品质提升、推动商业便民利民发展的项目;符合首都城市战略定位的促消费、稳增长等公共服务平台建设和典型示范类项目。对符合标准和要求的项目采取项目补助、政府购买服务、以奖代补等形式给予支持。
Next, we focused on DEPs in MII oocytes and zygotes and explored their important roles in the MZT process.There were 4,977 protein groups,which were obtained reliable quantitative data with at least two quantitative values in each group,used for further differential analysis.Notably,766 DEPs were identified with a 1.2-fold change in the cutoff and a statistical significance of P value < 0.05(Table S2).Moreover,498 DEPs were upregulated in 1-cell embryos,and 268 DEPs were upregulated in MII oocytes.With this differentially expressed proteome,we can distinguish the protein profiling and functional annotations between MII oocytes and 1-cell embryos(Figs. 4E and S6D). Many RNA- and ribosome-related functional pathways showed significant differences,consistent with the main task of the MZT phase.The pathway related to protein localization,which is related tothe accumulation of maternal material in oocytes,was mainly enriched in MII oocytes. However, the regulation of mRNA processing,peptide biosynthetic processes,cellular catabolic processes, spindle organization, organelle localization, and cytoskeleton organization were enriched in 1-cell embryos.These processes provide the materials and conditions for embryonic growth and development. Notably, the 1-cell embryo stage(P = 2.34 × 10-117) showed more significantly upregulated DEPs than the MII oocytes(P=9.16×10-35)did.
More interestingly,at the zygote stage,33 TFs(P=6.19×10-2),37 kinases (P = 1.28 × 10-4), and 2 core components of the SCMC(Figs. 4F and S6E) were significantly upregulated. This result suggests that numerous TFs, kinases, core SCMC members, and their associated protein-protein regulatory complexes, pathways, and networks may play critical roles during the very early stages of MZT.Therefore, the protein-protein interaction (PPI) networks for DEPs were constructed using the MIPPIE, TRRUST, and STRING datasets.In total, we found 1,146 nonredundant PPI relationships corresponding to 430 DEPs (Table S3). According to the significance of Pearson's correlation coefficient, we obtained 759 highly credible PPI relationships(P<0.05)corresponding to 373 DEPs.The core PPI network is presented in Fig. S7A. The Ywhae centered on the top four MCODE functional modules are shown in Fig. S7B, which are involved in translation, ribosomal subunits, DNA replication, and immune response. Considering the significantly regulated TFs and kinases, we deliberately selected trusted relationship pairs and mapped a detailed network corresponding to 99 nodes and 124 edges(Table S4).Fig.4G shows the core MCODE functional modules of these significantly regulated TFs and kinase-centered subnetworks, which are involved in the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)-protein kinase B (Akt) and Hippo signaling pathways and in the positive or negative regulation of stem cell population maintenance. Using pathway enrichment analysis(Fig.S7C),we observed that the cell cycle,TP53 regulation,estrogen signaling, and stress responses were regulated at both stages. More interestingly, lipid metabolism by peroxisome proliferators-activated receptor (PPAR) alpha and DNA-templated transcription initiation was significantly altered by the upregulated TFs and kinases at the MII stage, whereas tight junctions,insulin signaling, and Hippo signaling were altered by the upregulated TFs and kinases at the 1-cell stage. These results suggest that the significantly regulated TFs and kinases centered on PPI networks may be related to zygotic gene expression and zygotic protein regulators.
4. Discussion
Although proteomics is a well-established technique widely used in biological research, few studies on mouse oocyte meiotic maturation and pre-implantation embryo development have been published in the last decade because of the relatively large amount of starting material required. The development of single-cell or low-input multi-omics technologies has provided new hope in this field and promoted the understanding of transcriptional and translational regulation during mouse pre-implantation development [4,12,80]. However, single-cell or low-input proteomics remains in its infancy because of the properties of proteins such as complex constituents, low abundance, wide dynamic range, and lack of amplification ability.
In this study,we developed the CS-UPT for single-cell and lowinput mouse embryo samples.By using the deep coverage route,an average of 2,665 protein groups can be identified in a single mouse zygote,and 4,585 protein groups can be identified from 20 mouse zygotes,which has reached or exceeded the depth of the literature based on hundreds or thousands of mouse embryo samples[26-35]. We also established a high-throughput workflow for large-scale proteomic research. Based on TMT6plex labeling, the duration necessary for the preparation and MS analysis of 300 single mouse zygotes is 8.17 days,and an average of 2,182 or 2,371 protein groups can be identified from single zygotes with 50× or 100×carrier.On the basis of TMT8plex labeling,300 single mouse zygotes can be handled within 4.11 days. Remarkably, all these workflows are easy to use and automate. Without customized equipment, these ultrasensitive proteomic schemes can be conveniently promoted to other laboratories to enhance the comprehension of pre-implantation embryo development.Moreover,drug screening using high-throughput proteomics has shown great potential in various applications [81]. Vertebrate embryos are also widely used as tools for drug screening and pharmacological testing[82,83].Therefore,reducing the starting materials and highthroughput tools decreases the cost of drug screening and pharmacological testing, particularly for newly synthesized smallmolecule drugs.
The ZP is an extracellular coat of mouse oocytes and preimplantation embryos and is mainly composed of three proteins that play unique roles in reproduction and development.However,a high abundance of ZP proteins may interfere with the identification of other low-abundance proteins. Most proteomic studies have removed the ZP from oocytes or embryos, but few have investigated whether this removal step disturbs the proteome[30-35]. In this study, we not only demonstrated that our lowinput proteomic workflow performs well in oocytes or embryos with the ZP but also illustrated the influence of ZP removal in oocytes.Although most proteins were identified in both ZP-intact and ZP-free oocytes, and their expression levels did not change significantly, 151 proteins were significantly altered after ZP removal.Among them are some reproductive- or development-related proteins such as Apoa1 and Mfge8. To our knowledge, this study is the first to evaluate the influence of ZP removal from oocytes from a deep proteomic perspective.We hope that our results could serve as a reference for other researchers in related fields.
In this study, we used a low-input approach to obtain the first proteomic snapshot of the very early stages of mouse MZT.The indepth proteomic landscapes showed almost 5,500 protein groups and 766 quantitative differential proteomes from only 20 oocytes or 1-cell embryos per sample. Quantitative differences were observed in distinct proteome functional transformations at the very early stages of MZT. In this very early stage of MZT, we observed a fine regulatory network around 33 TFs,37 kinases,and two core components of SCMC. Notably, most of the differential expressed TFs,kinases,and SCMC members were upregulated in 1-cell embryos, suggesting that complex protein-protein regulatory network play a key role in minor ZGA at the 1-cell stage.
SCMC is emerging as a crucial maternally inherited biological structure during the initial stages of embryogenesis in mammals[84]. However, the overall integrated function of SCMC remains unclear, and the specific molecular mechanisms involved remain unresolved[84].Chen et al.[29]also used five components of SCMC(Ooep, Nlrp5, Tle6, Zbed3, and Padi6) detected by proteomics as “target proteins” to filter candidate maternal proteins based on the correlation of their expression with SCMCs. In this study, our proteome data precisely quantified almost all known SCMC members between MII oocytes and zygotes,including seven core components(Ooep, Zbed3, Nlrp5, Tle6, Khdc3, Padi6, and Nlrp4f) and three candidate SCMC members (Nlrp9a/b/c). Moreover, although the other SCMC members had not changed yet, Ooep and Zbed3 were significantly upregulated in the 1-cell stage, indicating that the dynamic expression regulation of key core components of SCMC might be initiated very early in the MZT.We concluded from the literature that Ooep is essential for oocyte cytoplasmic lattice formation and embryonic development at the maternal-zygotic stage transition[85]and that Zbed3 may be one of the downstream proteins mediating SCMC functions and regulating organelle distribution[86].
Overall, our findings provide an important resource for further investigations of the epigenetic,genomic,and proteomic regulation of early MZT events in mouse embryos.
5. Conclusions
In conclusion, we provided the CS-UPT for single-cell and lowinput mouse embryo samples. Both the deep coverage and highthroughput technology routes in our ultrasensitive proteomic platform will greatly improve proteomic research in the field.
Furthermore,we used a low-input proteomic approach to create the first large-scale snapshot for the very early stage of mouse MZT,providing new insights into this event in embryonic development.The significant protein regulatory networks centered on TFs and kinases between the MII oocyte and 1-cell embryo provide many insights into minor ZGA at the 1-cell stage.Moreover,the significant activation of several members of the kinase family involved in the insulin, Hippo,and PI3K-Akt signaling pathways suggests a potentially important role in the regulation of phosphorylation modification at the very early stage of MZT,which will encourage further research to explore the development and application of low-input phosphorylation modification technology in subsequent studies.
CRediT author statement
Lei GuandXumiao Li:Methodology, Validation, Formal analysis, Investigation, Data curation, Writing - Original draft preparation, Reviewing and Editing, Visualization;Wencheng Zhu:Methodology, Validation, Investigation, Data curation, Writing -Original draft preparation, Reviewing and Editing, Visualization,Funding acquisition;Yi Shen:Methodology, Validation, Software,Formal analysis, Investigation, Data curation, Writing - Reviewing and Editing, Visualization;Qinqin Wang:Formal analysis, Investigation, Visualization, Writing - Original draft preparation;Wenjun Liu:Formal analysis, Investigation, Validation, Writing -Reviewing and Editing;Junfeng Zhang:Data curation, Investigation, Validation, Writing - Reviewing and Editing;Huiping ZhangandJingquan Li:Data curation, Project administration, Writing -Reviewing and Editing;Ziyi Li:Conceptualization, Supervision,Resources, Software, Writing - Reviewing and Editing;Zhen Liu:Conceptualization, Supervision, Funding acquisition, Writing -Reviewing and Editing;Chen Li:Conceptualization, Supervision,Funding acquisition, Writing - Original draft preparation, Reviewing and Editing;Hui Wang:Conceptualization, Resources, Supervision, Funding acquisition, Writing - Reviewing and Editing.
Declaration of competing interest
Yi Shen, Junfeng Zhang, Huiping Zhang, and Ziyi Li are employees of Shanghai Applied Protein Technology Co.,Ltd.(Shanghai,China).The authors declare that there are no conflicts of interest.
Acknowledgments
We thank all members of our laboratories for helpful discussions. This work was supported by the National Natural Science Foundation of China(NSFC)(Grant Nos.:82030099 and 30700397),the National Key R&D Program of China (Grant No.:2022YFD2101500), the Science and Technology Commission of Shanghai Municipality, China (Grant No.: 22DZ2303000), the Shanghai Municipal Science and Technology Commission “Science and Technology Innovation Action Plan” Technical Standard Project,China (Grant No.: 21DZ2201700), the Shanghai Municipal Science and Technology Commission “Science and Technology Innovation Action Plan” Natural Science Foundation Project,China(Grant No.:23ZR1435800), the Strategic Priority Research Program of the Chinese Academy of Sciences, China (Grant No.: XDB32060000),the Basic Frontier Scientific Research Program of Chinese Academy of Sciences (Grant No.: ZDBS-LY-SM019), the Yangfan Project of Shanghai Science and Technology Commission, China (Grant No.:22YF1454100), and the Innovative Research Team of High-level Local Universities in Shanghai, China. We also thank the help from Dr. Chengpin Shen in Shanghai Omicsolution Co., Ltd. for database searching of partial mass spectrometry data.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2023.05.003.
猜你喜欢
中国水土保持(2021年1期)2021-01-15
行政事业资产与财务(2020年17期)2020-10-09
中国房地产·综合版(2018年3期)2018-05-23
新教育时代电子杂志(教师版)(2018年1期)2018-02-23
——河池至都安高速公路喀斯特地貌山区典型示范工程建设纪实
西部交通科技(2016年11期)2017-01-16
中国机电工业(2016年1期)2016-12-29
中国卫生(2016年8期)2016-11-12
课程教育研究·学法教法研究(2016年4期)2016-04-19
汽车观察(2016年3期)2016-02-28
红土地(2016年7期)2016-02-27
 Journal of Pharmaceutical Analysis2023年8期
Journal of Pharmaceutical Analysis2023年8期
- Journal of Pharmaceutical Analysis的其它文章
- Integrative multi-omics and systems bioinformatics in translational neuroscience: A data mining perspective
- Single-cell RNA sequencing reveals the dynamics of hepatic non-parenchymal cells in autoprotection against acetaminophen-induced hepatotoxicity
- Promise of spatially resolved omics for tumor research
- Temporal dynamics of microglia-astrocyte interaction in neuroprotective glial scar formation after intracerebral hemorrhage
- A single-cell landscape of triptolide-associated testicular toxicity in mice
- Single-cell and spatial heterogeneity landscapes of mature epicardial cells