
Single-cell RNA sequencing reveals the dynamics of hepatic non-parenchymal cells in autoprotection against acetaminophen-induced hepatotoxicity
2023-09-26 01:03LingqiYuJunYnYingqiZhnAnyoLiLinZhuJingyngQinFnfnZhouXioynLuXiohuiFn
Lingqi Yu , Jun Yn , Yingqi Zhn , Anyo Li ,b, Lin Zhu , Jingyng Qin ,b,Fnfn Zhou , Xioyn Lu ,b,c,e,f,*, Xiohui Fn ,b,c,e,g,**,
a Pharmaceutical Informatics Institute, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, 310058, China
b Future Health Laboratory, Innovation Center of Yangtze River Delta, Zhejiang University, Jiaxing, Zhejiang, 314100, China
c Innovation Center in Zhejiang University, State Key Laboratory of Component-Based Chinese Medicine, Hangzhou 310058, China
d School of Pharmacy, The University of Sydney, Sydney, 2006, Australia
e Jinhua Institute of Zhejiang University, Jinhua, Zhejiang, 321016, China
f Hangzhou Institute of Innovative Medicine, Zhejiang University, Hangzhou, 310058, China
g Engineering Research Center of Innovative Anticancer Drugs, Ministry of Education, Harbin,150023, China
Keywords:
Single-cell RNA sequencing
Drug-induced liver injury
Autoprotection against APAP hepatotoxicity
Endothelial cells
Dendritic cells
A B S T R A C T
Gaining a better understanding of autoprotection against drug-induced liver injury (DILI) may provide new strategies for its prevention and therapy.However,little is known about the underlying mechanisms of this phenomenon. We used single-cell RNA sequencing to characterize the dynamics and functions of hepatic non-parenchymal cells (NPCs) in autoprotection against DILI, using acetaminophen (APAP) as a model drug.Autoprotection was modeled through pretreatment with a mildly hepatotoxic dose of APAP in mice, followed by a higher dose in a secondary challenge. NPC subsets and dynamic changes were identified in the APAP (hepatotoxicity-sensitive) and APAP-resistant (hepatotoxicity-resistant) groups. A chemokine(C-C motif)ligand 2+endothelial cell subset almost disappeared in the APAP-resistant group,and an R-spondin 3+endothelial cell subset promoted hepatocyte proliferation and played an important role in APAP autoprotection. Moreover, the dendritic cell subset DC-3 may protect the liver from APAP hepatotoxicity by inducing low reactivity and suppressing the autoimmune response and occurrence of inflammation. DC-3 cells also promoted angiogenesis through crosstalk with endothelial cells via vascular endothelial growth factor-associated ligand-receptor pairs and facilitated liver tissue repair in the APAP-resistant group. In addition, the natural killer cell subsets NK-3 and NK-4 and the Sca-1-CD62L+ natural killer T cell subset may promote autoprotection through interferon-γ-dependent pathways.Furthermore,macrophage and neutrophil subpopulations with anti-inflammatory phenotypes promoted tolerance to APAP hepatotoxicity. Overall, this study reveals the dynamics of NPCs in the resistance to APAP hepatotoxicity and provides novel insights into the mechanism of autoprotection against DILI at a high resolution.
1. Introduction
Drug-induced liver injury (DILI) is a leading cause of drug development failure, restricted use, and market withdrawal. In Europe and the United States, DILI remains the leading cause of acute liver failure, and acetaminophen (APAP) use is typically associated with approximately 50%of acute liver failure cases in the United States and 30%-70% of cases in other western countries[1,2].The current foremost measure for the treatment of DILI is the immediate discontinuation of liver injury-associated drugs, followed by treatment with hepatoprotective, anticholestatic, or immunosuppressive drugs [3]. N-acetyl-L-cysteine is a clinically approved antidote for APAP hepatotoxicity, but it has limited efficacy in patients with advanced liver injury and induces adverse effects during long-term treatment[4].Corticosteroids are used as immunosuppressants in the allergic syndrome of DILI,but they can only elicit a response in a small fraction of drug-induced liver abnormalities [5]. Importantly, specific drugs for the treatment of DILI are still lacking because of the multigenic and complex pathogenesis of DILI. More efforts are needed to elucidate the mechanisms of liver injury and repair and discover relevant therapeutic targets for preventing DILI and associated liver failure.
Autoprotection is a phenomenon in DILI that manifests as resistance to re-exposure to the same toxicant. For instance, rodents repeatedly exposed to toxic doses of APAP develop resistance to higher doses in a secondary attack[6].Similar phenomena have been observed after re-exposure to carbon tetrachloride(CCl4)[7],thioacetamide [8], and 2-butoxyethanol [9]. The protective mechanisms underlying this phenomenon have attracted tremendous attention for their role in the prevention and treatment of DILI.Several studies have demonstrated that the regulation of cytochrome P450 and transporters, the induction of the unfolded protein response, the activation of Nrf2 and the detoxification gene Fmo3, and compensatory hepatocellular proliferation may be involved in protective adaptation[10-16].However,as liver injury and repair are complex processes involving multiple hepatic parenchymal and non-parenchymal cells(NPCs),further discovery of key cell populations and their biological functions in this protective adaptation is needed.
Single-cell RNA sequencing (scRNA-seq) has evolved into a powerful instrument for profiling highly heterogeneous cell populations in tissues. It allows for the discovery of key cell subsets during the autoprotective response with unsurpassed resolution and provides an additional dimension to bulk transcriptome data[17]. Recently, the kinetics of liver regeneration after acute APAP intoxication were investigated using scRNA-seq analysis, and a subset of hepatocytes that transiently upregulates fetal-specific genes to promote regeneration was identified [18]. After APAPinduced liver injury, many functional hepatocyte genes are transcriptionally replenished in non-proliferating hepatocytes to maintain essential functions[19].In addition to hepatocytes,NPCs,including hepatic stellate cells (HSCs), macrophages, and endothelial cells, play important roles in immune recruitment, proliferation,and matrix remodeling[18].Endothelial cells play a role in liver regeneration after APAP overdose via wingless-type mouse mammary tumor virus (MMTV) integration site family member 2 and 9b (Wnt2 and Wnt9b) signaling [20]. Macrophage-derived Wnts are important for cell proliferation after APAP hepatotoxicity[19].However,these studies have focused on the mechanisms of liver repair after a single exposure to APAP, which may be quite different from the mode of action by which autoprotection occurs.Additionally, the role of NPCs in the adaptation response to APAPinduced liver injury and the interaction between different types of cells require further study.
With a special focus on NPCs, this study aimed to identify key cell subsets that serve a vital function in the repair of and resistance to APAP hepatotoxicity. Liver NPC suspensions from single-dose APAP-treated and secondary attack-exposed mice were collected and subjected to scRNA-seq analysis to reveal the dynamic changes and functions of NPCs and identify subpopulations with different roles in autoprotection against APAP hepatotoxicity.
2. Methods and materials
2.1. Mouse model
Male C57BL/6 mice weighing 18-20 g were obtained from Zhejiang Vital River Laboratory Animal Technology Co., Ltd. (Jiaxing,China)and housed under constant conditions in a 12:12 h lightdark cycle at 24±2°C.Animal experiments were approved by the Animal Care and Use Committee of Zhejiang University School of Medicine (Approval number: ZJU20230134). Mice were acclimatized for 3 day before the start of the experiments and then randomly divided into three groups: the control group (n = 8),APAP group (n = 9), and APAP-resistant group (n = 8). After overnight fasting, the mice in the APAP-resistant group were administered 300 mg/kg of APAP by intraperitoneal injection, and mice in the APAP and control groups were injected with the same volume of saline at 0.1 mL/10 g.Two days later,mice in the APAP and APAPresistant groups were intraperitoneally administered APAP at a dosage of 600 mg/kg, and mice in the control group were administered the same volume of saline(0.1 mL/10 g).Twenty-four hours after the second exposure, mice were anesthetized with 1.5% sodium pentobarbital. Blood samples were then collected and centrifuged at 4,000 rpm for 10 min at 4°C(5810R;Eppendorf Ltd.,Hamburg, Germany). Serum was stored at -80°C until the next step of the experiment.Mouse livers were removed,and three from each group were mixed for scRNA-seq analysis. The remaining livers were fixed in 10% neutral buffered formalin for further histopathological examination.
2.2. Biochemical and histopathological evaluation
The activity of serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) was measured with a Cobas C8000 system (Roche Diagnostics Ltd., Mannheim,Germany).
Mouse liver tissues fixed in 10%neutral buffered formalin were embedded in paraffin after dehydration and cut into 4-μm-thick slices. The tissue was stained with hematoxylin and eosin and inspected under a light microscope (OLYMPUS IX53, Olympus Corporation Inc., Tokyo, Japan).
2.3. Single-cell dissociation from liver samples
Single-cell suspensions from mouse livers were prepared using a Mouse Liver Dissociation Kit (Miltenyi Biotech Inc., Bergisch Gladbach, Germany) according to the manufacturers' protocols.Briefly, mouse livers were enzymatically dissociated with the gentleMACS™Dissociator (Miltenyi Biotech Inc.). After dissociation,samples were passed through a 40 μm filter to remove larger particles, and dead cells were then removed using a Dead Cell Removal Kit (Miltenyi Biotech Inc.). The Mouse Liver Dissociation Kit is optimized to obtain a high yield of non-parenchymal mouse liver cells. Therefore, non-parenchymal mouse liver cells were obtained and used for scRNA-seq analysis.
2.4. Preparation and sequencing of single-cell libraries
The suspensions of liver cells were encapsulated into droplets using a Single Cell 3'Reagent Kit v2(10×Genomics Inc.,Pleasanton,CA, USA). After lysis, reverse transcription was performed using a thermal cycler (ProFlex PCR, Thermo Fisher Scientific Inc., Waltham, MA, USA), followed by purification and amplification with Dynabeads for 10 or 14 cycles.The amplified cDNA was fragmented and ligated with connectors and sample indexes. The resulting fragments were then selected using solid-phase reversible immobilization beads(Beckman Coulter Inc.,Brea,CA,USA).The libraries were built and sequenced on a NovaSeq 6000 Illumina sequencing platform (Illumina, San Diego, CA, USA).
2.5. Quantification, quality control, and clustering analysis of single-cell RNA expression
Raw sequencing data obtained from 10× Genomics were matched and quantified using the CellRanger suite (3.0.2,10×Genomics Inc.).Using the mouse genome as a reference,reads were mapped by the CellRanger count module. The Seurat R package(version 3.1.5)was used to exclude cells that expressed less than 200 or more than 5,000 unique genes,or those with more than 10%of the reads mapped to mitochondria were excluded.The gene expression matrix of the remaining cells was normalized using global scaling. Highly variable genes were found using the “Find-VariableFeatures” function in the Seurat R package,and all samples were integrated together with the “IntegrateData” function. The “ScaleData” function was chosen to scale the integrated data.
Principal component analysis was performed using runPCA,and t-distributed stochastic neighbor embedding (t-SNE) was conducted using the first 30 principal components to embed the dataset into two dimensions. The “FindNeighbors” and “FindClusters” functions were then used to build nearest neighbor graphs and clustering units,respectively.The known markers were used to annotate the cell types of each cluster and a very low percentage of hepatocytes was removed. Principal component analysis was performed on each type of NPC,and the first 15 principal components were selected for clustering to obtain cell subclusters.
2.6. Gene Ontology (GO) enrichment analysis
Differentially expressed genes (DEGs) between two clusters were obtained using the “FindAllMarkers” function with the parameters “only.pos” set to “TRUE”, “min.pct” set to 0.25, and “logfc.threshold” set to 0.5. The genes with P adjust < 0.05 were retained. The upregulated DEGs of these clusters were then analyzed using the “clusterProfiler” R package for GO enrichment[21]. A significance level of P < 0.05 was used to determine the enrichment results.The GO enrichment analysis was filtered based on the q value (“qvalueCutoff” = 0.2).
2.7. Pseudotime analysis
Pseudotime-series analysis was performed using the R language package Monocle (version 2.22.0) [22]. Monocle combines the unsupervised data-driven method with the inverse graph embedding to accurately reconstruct single-cell trajectories.
2.8. Cell-cell communications analysis
CellPhoneDB[23]is a tool to analyze cell-cell communication by gene expression and communication information from known databases. Any gene that was expressed in less than 10 cells was filtered out.The filtered gene expression matrix was then imported into CellPhoneDB(version 3.1.0)and analyzed.Interacting values of ligand-receptor pairs without a valid mean (mean > 0.5) were filtered out.
2.9. Cell culture
Human hepatocellular carcinoma (HepG2) cells (Kunming Cell Bank of Type Culture Collection, Chinese Academy of Sciences,Kunming, China) and human umbilical vein endothelial cells(HUVECs)(Chinese Academy of Sciences Typical Culture Collection Committee Cell Bank, Shanghai, China) were cultured in highglucose Dulbecco's modified Eagle's medium (DMEM, Gibco,ThermoFisher Scientific Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (Gibco, ThermoFisher Scientific) and 1%antibiotic-antimycotic solution(Gibco,ThermoFisher Scientific).Both cells were maintained in an incubator at 37°C with a 5%CO2atmosphere. The medium was changed every 2 day. Cells were passaged at 80% confluency and used at 3-20 generations.
2.10. Cell transfection, co-culture, and proof-of-function
HUVECs were cultured in 6-well flat-bottom plates at a density of 2.8 × 105cells/well, and the culture medium was replaced with fresh DMEM complete medium after 24 h. Then, 5 μL of Lipofectamine 8000(Beyotime Biotechnology Inc.,Shanghai,China),125 μL of OptiMEM(Thermo Fisher Scientific),and 2.5 μg of plasmid pEX-3 with or without specific gene cDNA (GenePharma Ltd., Shanghai,China)were mixed homogeneously,left for 5 min,and added to the 6-well flat-bottom plates.After 4 h of incubation,the medium was replaced with fresh DMEM complete medium. The culture supernatant of HUVECs was collected after 48 h of incubation, during which time the plasmid showed good transfection efficiency in HUVECs.
The HepG2 cells were cultured in 12-well,flat-bottom plates at a density of 2 × 105cells/well. After 24 h of incubation, the culture medium was replaced with the supernatants collected from HUVECs, and 20 mM APAP was added. The culture supernatant of another empty vector plasmid,pEX-3,was used as the control.After 24 h, the viability of HepG2 cells was examined using the Cell Counting Kit-8 assay.The absorbance of the solution was measured at 450 nm using an Infinite M1000 Pro spectrophotometer(TECAN Ltd., M¨annedorf, Germany). The absorbance of the empty vector plasmid pEX-3 control was set as 100% cell viability.
2.11. High-performance liquid chromatography (HPLC)determination of plasma concentrations of APAP in mice
After 3 day of acclimatized feeding, C57BL/6 mice were randomly divided into the control, APAP, and APAP-resistant groups. Each group was divided into three time periods: 3(n=3),6(n=3),and 24 h(n=5).The specific APAP administration and procedure were the same as that in Section 2.1. Mice were anesthetized with 1.5% pentobarbital sodium at 3 h, 6 h, and 24 h after the second exposure.Blood samples were then collected from the inferior vena cava into anticoagulation tubes,placed on ice,and centrifuged at 4,000 rpm for 10 min at 4°C(5810R,Eppendorf).The obtained plasma was stored at -80°C until the next step of the experiment.
For every 100 μL mouse plasma,1 mL of 2-propanol:chloroform(5:95,V/V)was added,vortexed for 1 min,and then centrifuged at 4,500 rpm for 10 min. The organic phase layer was carefully separated and concentrated by centrifugation under reduced pressure until the liquid was completely evaporated.The tube containing the dried residue was added with 50 μL mobile phase (water:-methanol:formic acid,70:30:0.15,V/V/V),vortexed for 30 min,and then centrifuged at 12,000 rpm for 10 min.The upper layer of liquid was carefully removed for the subsequent chromatographic analysis. Chromatographic analysis was performed using an Agilent 1260 HPLC system(Agilent Technologies Inc.,Santa Clara,CA,USA)with a C18reversed-phase column(250 mm×3.9 mm,5 μm).The flow rate of the mobile phase was set to 0.8 mL/min and the column temperature was set at 30°C.Detection was performed at 254 nm with 10 μL of plasma extract per injection.
2.12. Statistical analysis
Data are presented as the mean ± standard deviation. An unpaired two-sided t-test was used to detect statistical significance between the groups. P < 0.05 was considered statistically significant. ScRNA-seq analyses and graph generation were conducted using R (version 4.1.3).
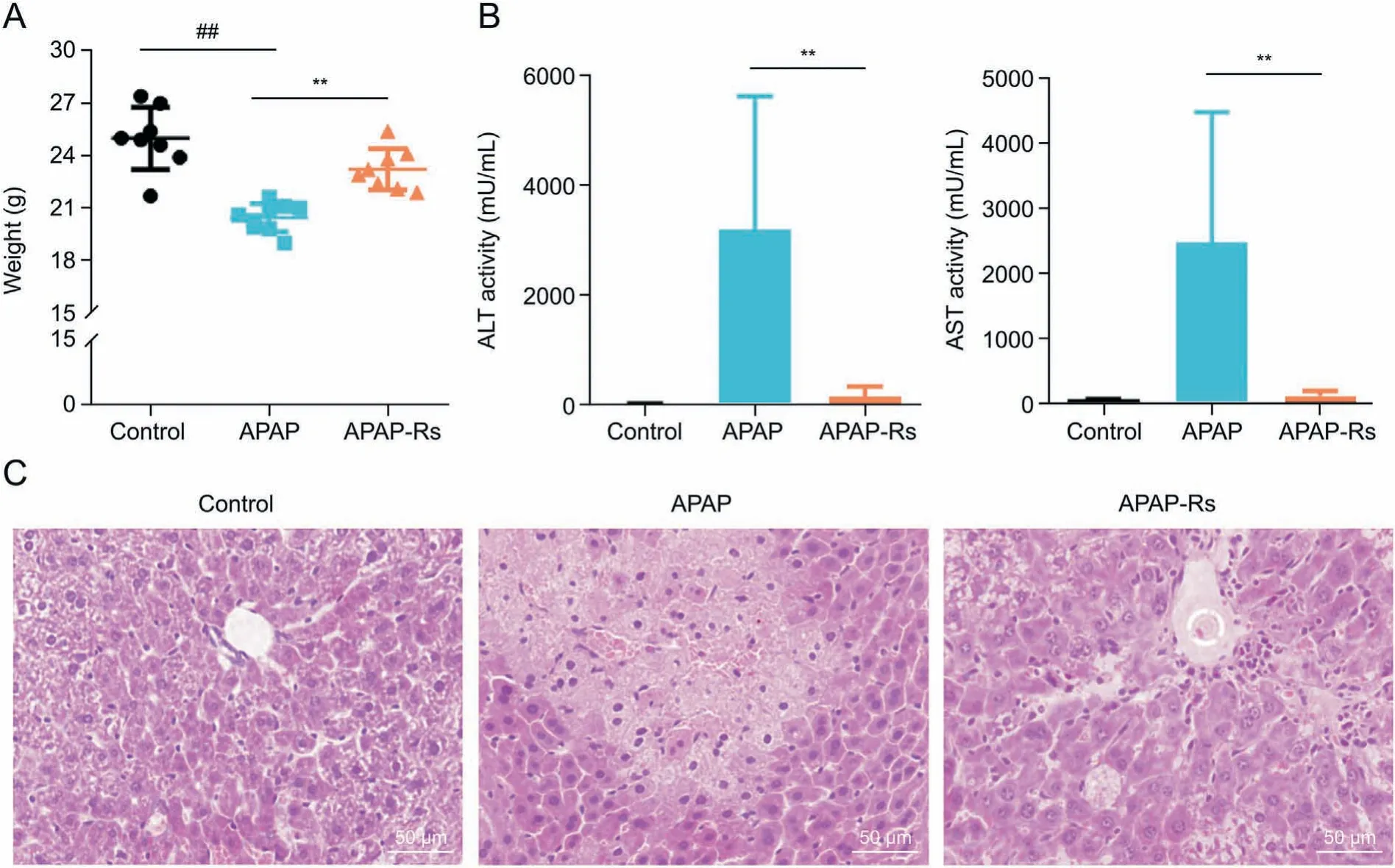
Fig. 1. Acetaminophen (APAP) pretreatment significantly decreased APAP hepatotoxicity. (A, B) Body weight (A) and serum biochemical parameters (B) of mice in the control(n=8),APAP(n=9),and APAP-resistant(n=8)groups.Data are shown as the mean±standard deviation(SD). ##P<0.01,compared with the control group; **P<0.01,compared with the APAP group. (C) Hematoxylin and eosin staining of liver tissues after APAP administration. Original magnification × 400. APAP-Rs: APAP-resistant group; ALT: alanine aminotransferase; AST: aspartate aminotransferase.
3. Results
3.1. Protection of mice from APAP hepatotoxicity by APAP pretreatment
Previous studies have reported the phenomenon of autoprotection against APAP hepatotoxicity [13,14]. In this study, the APAP autoprotection model was established according to a previously described model [13,14]. Briefly, mice were pretreated with a low dose of APAP(300 mg/kg),followed by a challenge with a high dose of APAP (600 mg/kg) 2 day later. The serum levels of ALT and AST were utilized as biochemical markers to assess liver injury, along with other indicators like histopathological examination abnormalities and loss of body weight. Compared with that in the control group,the APAP group showed a significant decrease in body weight,accompanied by a marked increase in serum ALTand AST levels(Figs.1A and B).Consistent with these indicators,extensive hepatocellular damage was observed in the livers of mice in the APAP group. In contrast,the body weight,serum biochemical,and histopathological examinations of the APAP-resistant group tended to normalize,except for a slight hydropic degeneration observed in the liver(Fig.1C).Death was not observed in any treatment group during the study period.In addition,24 h after the challenge administration,the plasma concentrations of APAP were found to be lower in mice pretreated with APAP (Fig. S1), which further supported the hypothesis that APAP pretreatment has a protective effect.
3.2. Single-cell transcriptional profiling of hepatic NPCs in autoprotection against APAP hepatotoxicity
To explore the mechanisms underlying autoprotection and identify the key NPCs involved in this response, scRNA-seq analysis was conducted on NPCs collected from the APAP and APAP-resistant groups using the 10× Genomics platform(Fig. 2A). After quality filtering, a total of 33,306 cells were detected, of which 14,550 and 18,756 cells were obtained from the APAP-resistant and APAP groups, respectively. After unsupervised clustering and t-SNE dimensionality reduction, nine different cell types were identified according to their marker gene expressions (Figs. 2B-D), including B cells (5398), dendritic cells (DCs; 841), dividing cells (543), endothelial cells (4229),HSCs (46), macrophages (2817), neutrophils (6185), T cells(9404), and natural killer (NK) cells (3843). Before and after eliminating the influence of the cell cycle, the t-SNE analysis results showed no significant changes, indicating that the cell cycle had no significant influence on the clustering of hepatocyte data (Fig. S2). As shown in Fig. 2C, the proportion of these cell lineages varied greatly between the APAP and APAP-resistant groups, indicating a heterogeneous NPC status. It is worth noting that, compared with those in the APAP group, the proportions of dividing cells and DCs were increased in the APAPresistant group, while the proportion of neutrophils was decreased.
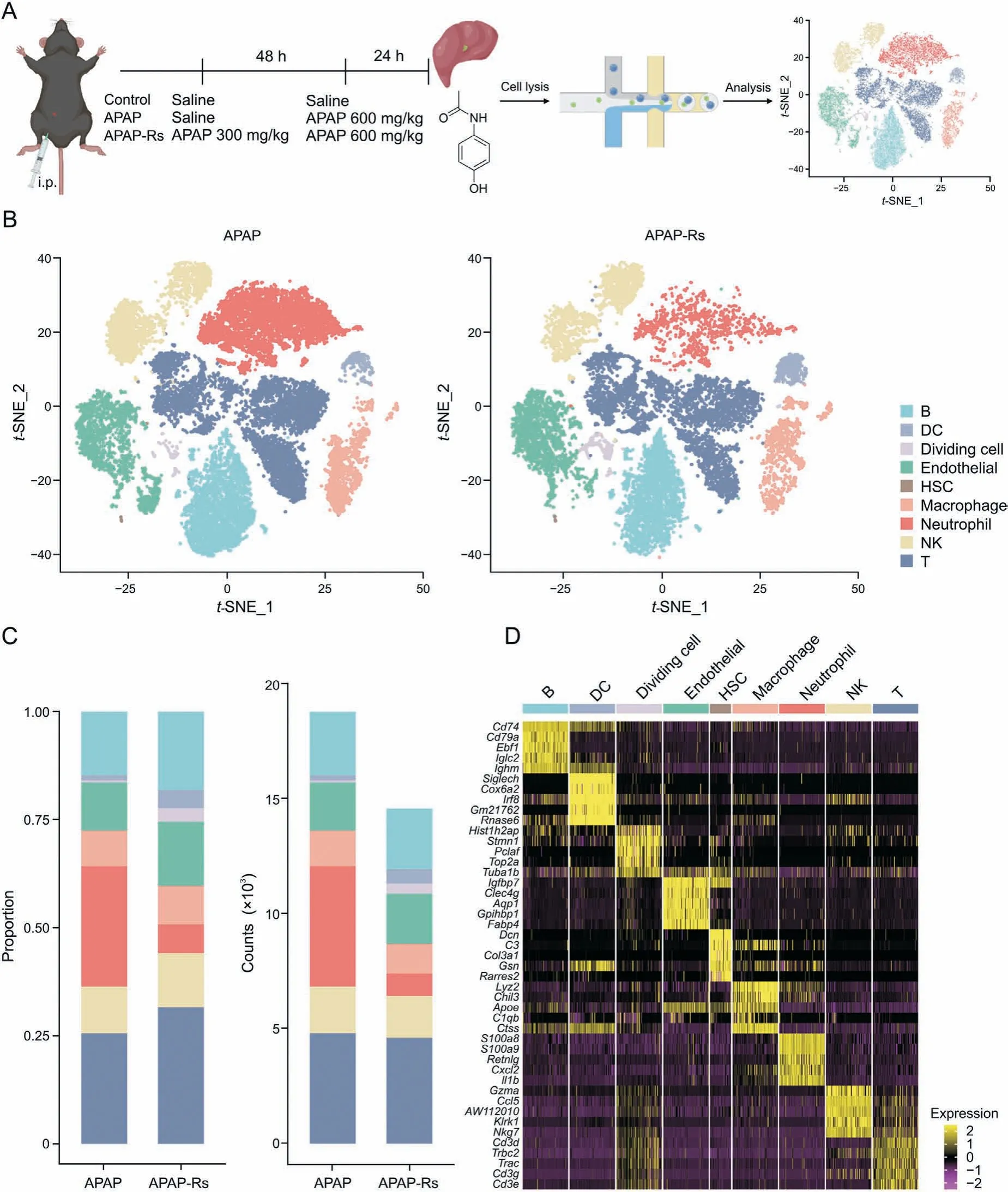
Fig. 2. Single-cell transcriptome changes in hepatic non-parenchymal cells (NPCs) for resistance to acetaminophen (APAP) hepatotoxicity. (A) Flow diagram of single-cell RNA sequencing experiments. Liver tissues from three representative mice were collected from each of the APAP and APAP-resistant groups. (B) The t-distributed stochastic neighbor embedding(t-SNE)projections of NPCs in mouse livers in the APAP(left)and APAP-resistant(right)groups.(C)Proportions(left)and numbers(right)of nine cell types in the APAP and APAP-resistant groups.(D)Heatmap of the relative expression of marker genes in different types of liver non-parenchymal cells.APAP-Rs:APAP-resistant group;DC:dendritic cell; HSC: hepatic stellate cell; NK: natural killer.
3.3. R-spondin 3 (Rspo3)-positive subpopulations of endothelial cells are responsible for tolerance to APAP hepatotoxicity
The subpopulations of endothelial cells in the APAP-resistant group were obviously changed compared with those in the APAP group. To further clarify the role of endothelial cells in autoprotection against APAP-induced liver injury, we performed a second-level cluster analysis of endothelial cells and identified four subpopulations (Fig. 3A): chemokine (C-C motif) ligand 2+(Ccl2+)cells,pericentral endothelial cells(Endo-pc),periportal endothelial cells (Endo-pp), and liver sinusoidal endothelial cells (LSECs) [24].Transcriptional heterogeneity of endothelial cells was detected in both the APAP and APAP-resistant group (Fig. 3B). The Ccl2+subpopulation was almost eliminated in the APAP-resistant group after APAP pretreatment, while the Endo-pc and Endo-pp subpopulations increased in both number and proportion compared with those in the APAP group.
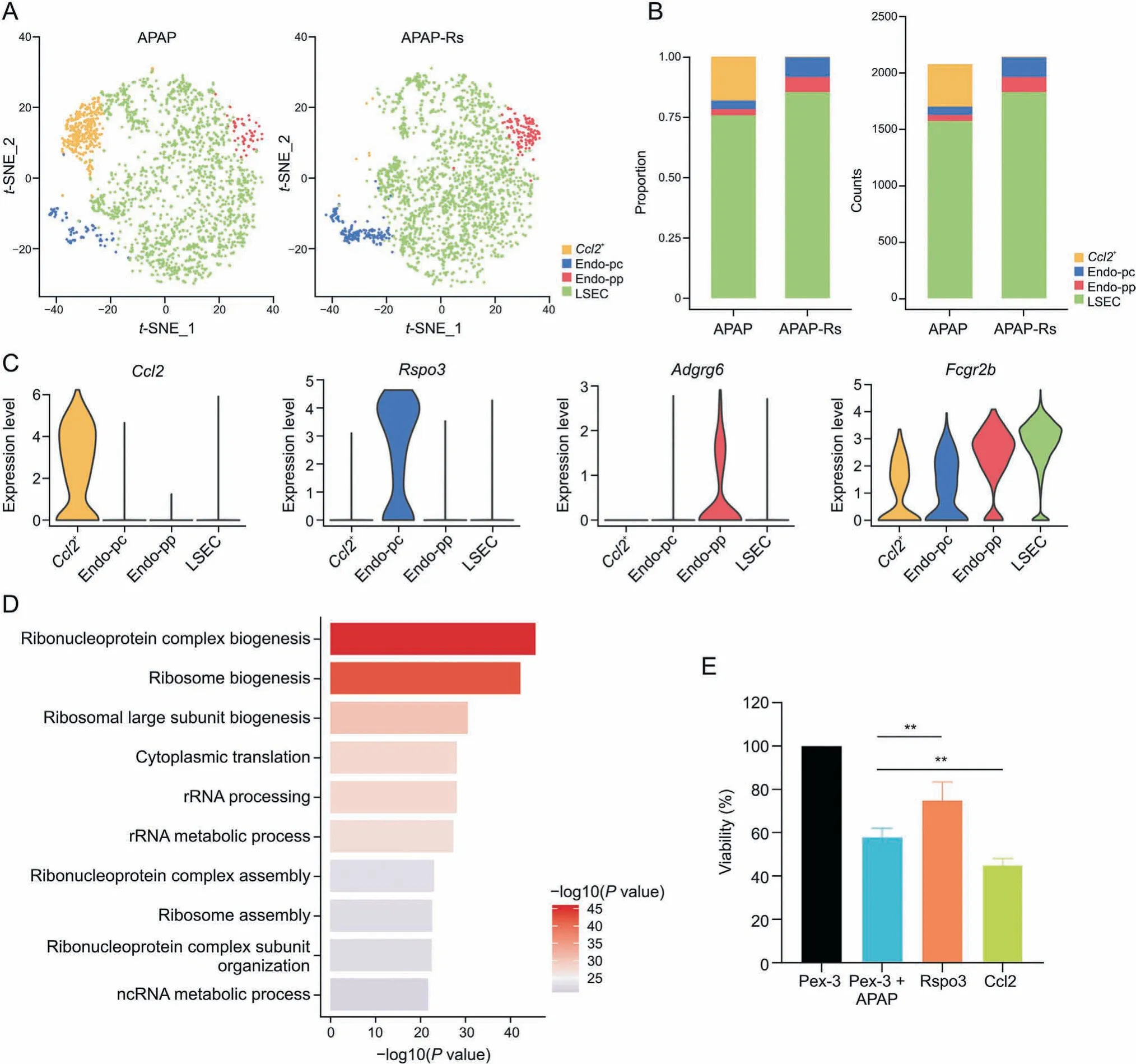
Fig. 3. R-spondin 3 (Rspo3)-positive subpopulations of endothelial cells are important for acetaminophen (APAP) autoprotection. (A) The t-distributed stochastic neighbor embedding(t-SNE) projections of endothelial cell subpopulations in the livers of APAP(left)and APAP-resistant(right) mice.(B)Proportions(left)and numbers(right) of the four subpopulations in the endothelial cells. (C) Violin plots of the expression levels of marker genes in the endothelial cell subpopulation. (D) Gene Ontology enrichment terms of chemokine (C-C motif)ligand 2+ (Ccl2+) endothelial cells.(E) Viability of human hepatocellular carcinoma cells in co-culture experiment. **P< 0.01, compared with other groups.APAP-Rs: APAP-resistant group. LSEC: liver sinusoidal endothelial cells; Endo-pc: pericentral endothelial cells; Endo-pp: periportal endothelial cells; Adgrg6: adhesion G proteincoupled receptor G6; Fcgr2b: Fc receptor IgG low affinity 2b.
Fig. 3C shows that only the Ccl2+subpopulation expressed the chemokine ligand Ccl2, which was barely expressed in the other subpopulations, and that only Endo-pc highly expressed Rspo3.Pathway analysis revealed that ribosome-related pathways were significantly enriched in the Ccl2+subgroup based on DEGs between this cluster and other clusters, including ribosome biogenesis,rRNA metabolic process,and ribosome assembly(Fig.3D).The monocyte chemoattractant protein-1 (also known as CCL2) is involved in the migration and infiltration of monocytes and macrophages[25],and can be induced by a variety of mediators,such as interleukin-1 beta(IL-1β)and tumor necrosis factor-alpha(TNF-α),to respond to the pro-inflammatory response [26-28]. The near disappearance of this subpopulation suggested that APAP pretreatment suppressed the inflammatory response in the APAPresistant group compared to that in the APAP group. The Endo-pc subcluster highly expressed Rspo3, which encodes a protein that plays an important role in the regulation of the Wnt/β-catenin signaling pathway. This pathway contributes to liver regeneration and protects the liver from oxidative damage by preserving mitochondrial function [29,30]. Consistently, the abundance of this subpopulation increased after APAP pretreatment (Fig. 3B).Pathway enrichment analysis showed that the negative regulation of phosphate metabolic processes, blood coagulation, and hemostasis were strongly activated in Endo-pc.
To detect the effects of these subclusters on APAP hepatotoxicity,we transfected HUVECs (an endothelial cell line) with overexpression plasmids, resulting in high Rspo3 or Ccl2 expression.Cells were then co-cultured with APAP-treated HepG2 cells to evaluate the effect of Rspo3 and Ccl2 on APAP-induced liver injury.The results showed that high Rspo3 expression in endothelial cells could protect against APAP hepatotoxicity, while high Ccl2 expression in endothelial cells had the opposite effect (Fig. 3E).
3.4. DC subpopulations facilitate autoprotection against APAP hepatotoxicity by suppressing the autoimmune response and promoting angiogenesis
Since DCs are a major determinant of intrahepatic immunity and exhibit heterogeneity [31,32], we further explored their subsets by unsupervised clustering.Three subpopulations,named DC-1-3, were identified (Figs. 4A, S3A, and S3B). They all highly expressed Siglec-H and lost the expression of Itgam (Fig. S3C),indicating that they are plasmacytoid dendritic cells (pDCs) [33].Although hepatic DCs usually mediate immune tolerance rather than immunogenicity [31], they can become pro-inflammatory and induce secondary hepatic injury after APAP exposure [34].Consistently, DC-1 and -2 cells from the APAP group showed higher Tnf, Ccl2, Tlr2, Tlr4, Tlr7, and/or Tlr9 expression levels(Fig. 4B) than those from the APAP-resistant group. Moreover, a large number of DCs appeared in the APAP-resistant group, which was consistent with the observation that DC depletion aggravated APAP-induced liver injury [31].
Compared with that in the APAP group, the proportion of DC-1 cells decreased in the APAP-resistant group, while both the proportion and number of DC-3 cells increased(Fig.4C).GO enrichment and metabolic process enrichment analysis showed that metabolism-related pathways were enriched in the DC-1 subpopulation (Figs. 4D and E), including glycolytic processes, adenosine triphosphate (ATP) generation from adenosine diphosphate (ADP),ADP metabolic processes, glucose metabolism, glycan metabolism,and lipid metabolism. Moreover, the metabolic score of DC-1 cells was significantly higher than that of the DC-2 and-3 subsets(Fig.4F).After the occurrence of APAP-induced liver injury, the increased numbers of DC-1 cells may respond to the stimulation of liver injury through the energy metabolism pathway, whereas liver damage in the APAP-resistant group was mild, and the numbers of DC-1 cells were relatively decreased. The pseudotime analyses for trajectory showed that DC-2 cells exhibited intermediate state characteristics between DC-1 and DC-3 cells (Fig. 4G). DC-3 cells specifically expressed mitochondrial genes, including mt-Co1, mt-Nd4, mt-Co2,mt-Cytb, mt-Co3, mt-Nd1, and mt-Atp6. It indicated that autoprotection against APAP hepatotoxicity may be related to the promotion of mitochondrial metabolic pathways in DCs(Fig.4H).The metabolic pathways highly active in DC-3 cells were different from those in DC-1 and-2 cells,in which oxidative phosphorylation and other related metabolic pathways were enriched (Fig. 4E). Tolerogenic DCs enhance the metabolic pathway of oxidative phosphorylation and facilitate the oxidation of fatty acids[35].The evidence suggests that DC-3 cells may be a subset of tolerogenic DCs that play a role in selfrecognition and exhibit low reactivity.
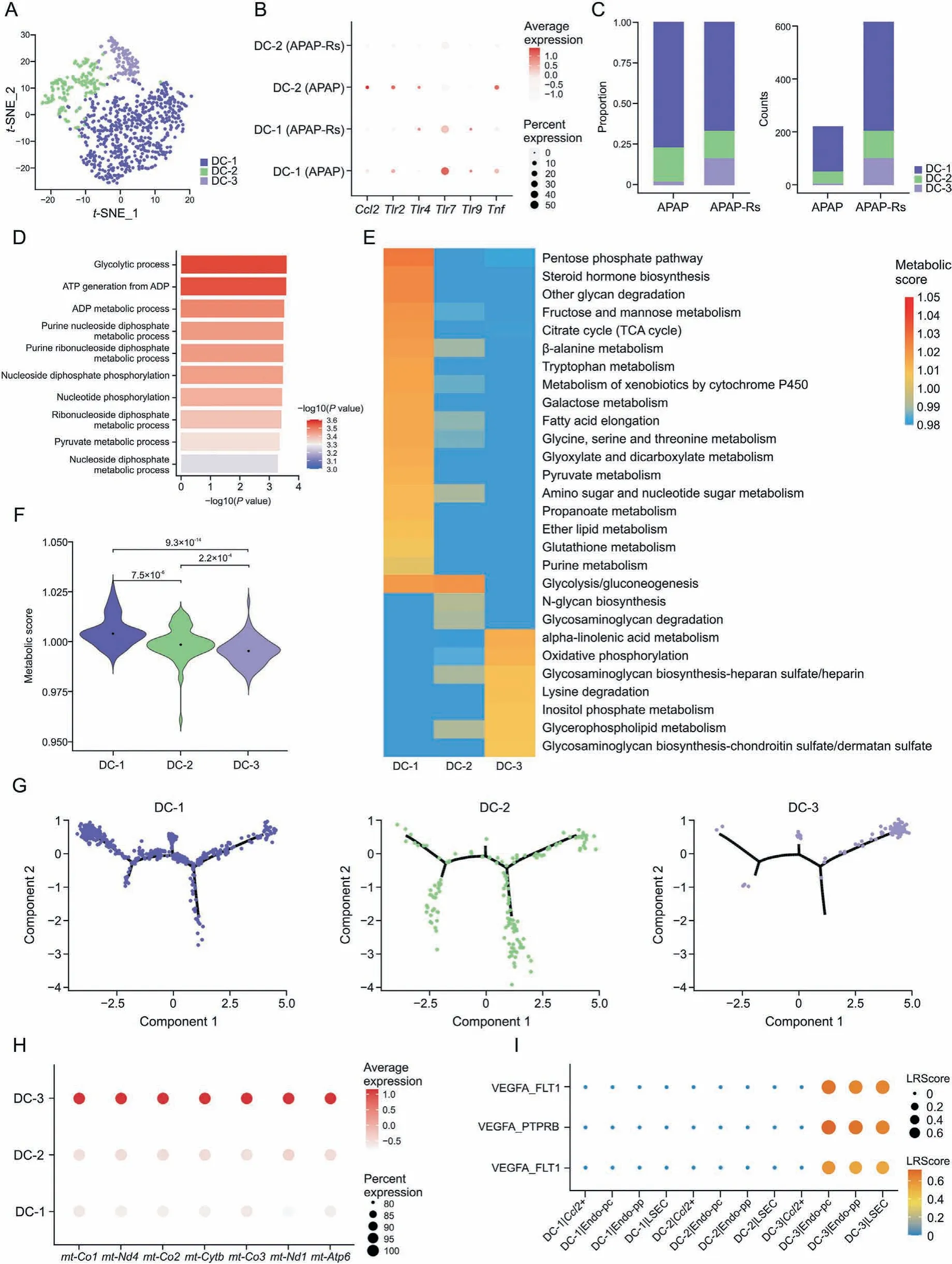
Fig.4. Dendritic cell(DC)subpopulations facilitate autoprotection to acetaminophen(APAP)hepatotoxicity.(A)The t-distributed stochastic neighbor embedding(t-SNE)plot of 841 DCs shows three clusters labeled as DC-1,DC-2,and DC-3.(B)Dot plots of Tnf,Ccl2,Tlr2,Tlr4,Tlr7,and Tlr9 in DCs.(C)Fraction of each DC subset relative to all DCs(left)and the cell quantity of each subset(right).(D)Gene Ontology enrichment terms of DC-1.(E)Metabolic process enrichment terms of DC subsets.(F)A violin plot showing the metabolic score in each DC subpopulation.(G)Trajectory analysis including DC-1-3.(H)Dot plots of mt-Co1,mt-Nd4,mt-Co2,mt-Cytb,mt-Co3,mt-Nd1,and mt-Atp6 in DCs.(I)Bubble plots exhibiting significant interactions between DCs and endothelial cells by the ligand-receptor pairs vascular endothelial growth factor B (VEGFB)/fms related tyrosine kinase 1(FLT1), vascular endothelial growth factor A(VEGFA)/protein tyrosine phosphatase receptor type B(PTPRB),and VEGFA/FLT1.APAP-Rs:APAP-resistant group.LSEC:liver sinusoidal endothelial cells;Endo-pc: pericentral endothelial cells; Endo-pp: periportal endothelial cells.
CellTalkDB was used to explore the crosstalk between subpopulations of DCs and endothelial cells, and the results showed that DC-3 cells interacted with Endo-pc, Endo-pp, and LSECs through vascular endothelial growth factor B (VEGFB)/fms related tyrosine kinase 1 (FLT1), VEGFA/protein tyrosine phosphatase receptor type B (PTPRB), and VEGFA/FLT1 ligand-receptor pairs, but not with the Ccl2+subset(Fig.4I).DCs produce and release various proangiogenic and antiangiogenic mediators according to their activation state and cytokine environment, mediating proangiogenic activity[36].Therefore,DC-3 cells promoted the angiogenesis of endothelial cells through VEGF-associated ligand-receptor pairs,promoting liver injury repair in the APAP-resistant group.
3.5. NK subpopulations promote autoprotection against APAP hepatotoxicity through interferon-γ (IFN-γ) pathways
NK cells are abundant in the liver,especially compared with that in peripheral lymphoid organs [37]. t-SNE visualization revealed four subclusters of NK cells,termed NK-1-4,which were identified based on their highly expressed genes(Fig.5A).Both the proportion and number of NK-1 and NK-2 cells were decreased in the APAPresistant group, while those of NK-3 and NK-4 cells were increased compared with those in the APAP group (Fig. 5B). NK-1 and NK-2 cells expressed high levels of inflammatory and chemotactic genes, including Tnfrsf9, Tnfrsf18, S100a1, Cxcr3, and Cxcr6(Fig. 5C). GO enrichment analysis also showed that NK-1 and NK-2 cells were responsible for activating the immune response and leukocyte migration,indicating that the inflammatory response was activated after APAP-induced liver injury. Killer cell lectin-like receptor(KLR)genes(Klra4,Klra8,Klra7,Klra3,Klra9,and Klrg1)were abundantly expressed in NK-3 and NK-4 cells (Fig. 5C).The activation of KLRs in NK cells is associated with the expression of IFN-γ[38],and IFN-γ production was the top enrichment pathway based on the DEGs of NK-3 and NK-4 cells(Fig.5D).NK cells are known to produce IFN-γ,which can promote the regeneration of hepatocytes derived from bone marrow in liver diseases such as glucose-6-phosphatase catalytic subunit deficiency, CCl4treatment, or 2-acetylaminofluorene/partial hepatectomy-induced liver injury,thereby promoting liver repopulation[39].Hepatic NK cells stimulate the production of IL-6 in Kupffer cells through IFN-γ,resulting in the inhibition of cholestatic liver injury[40].Thus,promoting NK-3 and NK-4 cells may be one of the mechanisms involved in adaptation to APAP hepatotoxicity.Furthermore,NK cells can also inhibit liver fibrosis by IFN-γ production,which leads to HSC apoptosis and cell cycle arrest in a signal transducer and activator of transcription 1 dependent manner[41,42],implying that NK-3 and NK-4 cells may also be involved in the prevention of liver fibrosis.
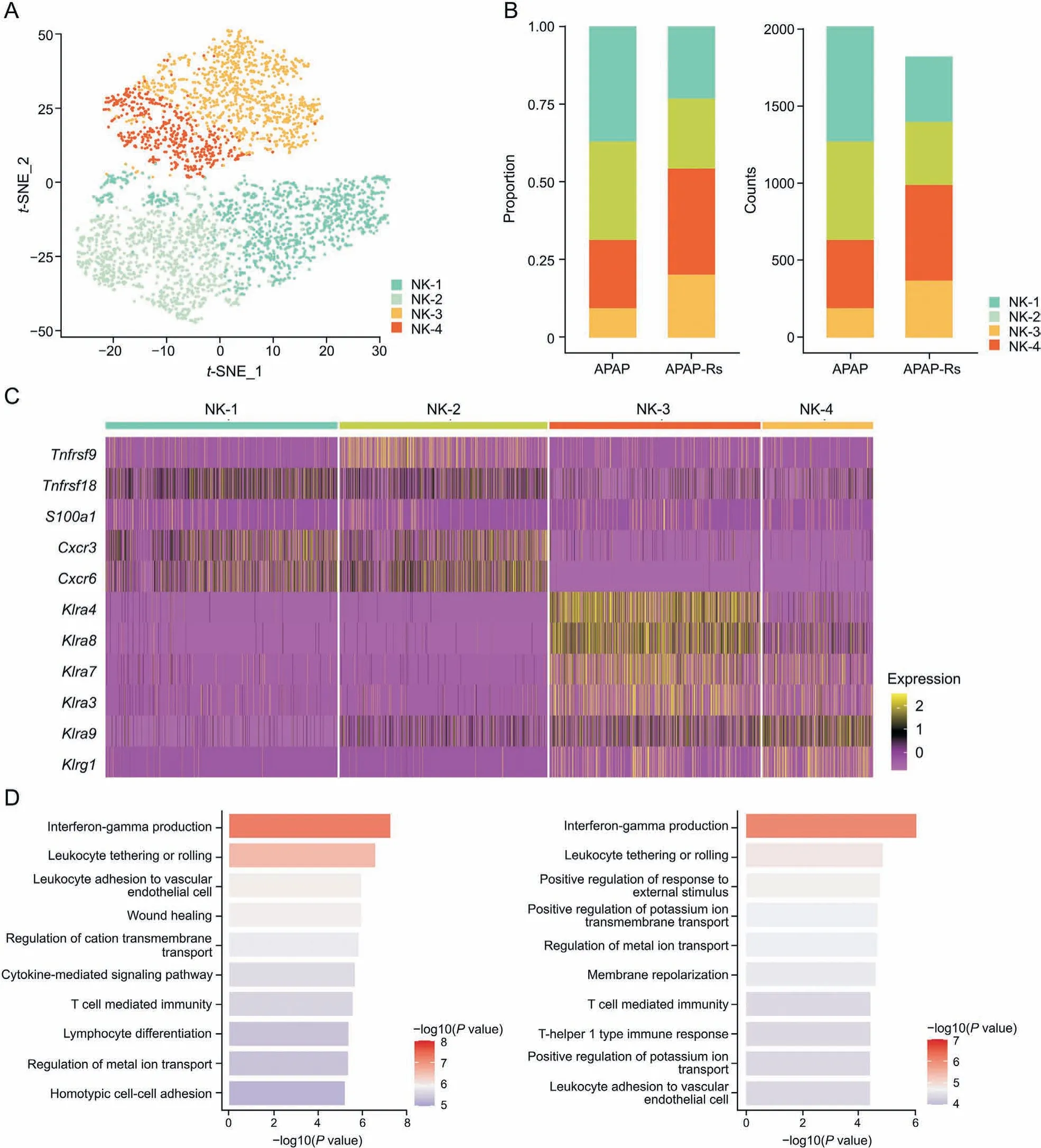
Fig. 5. Natural killer (NK) subpopulations promote autoprotection to acetaminophen (APAP) hepatotoxicity. (A) The t-distributed stochastic neighbor embedding (t-SNE) plot of 3843 NK cells shows four clusters labeled NK-1-4. (B) Fraction of each NK subset relative to all NK cells (left) and the cell quantity of each subset (right). (C) Heatmap of Tnfrsf9,Tnfrsf18,S100a1,Cxcr3,Cxcr6,Klra4,Klra8,Klra7,Klra3,Klra9,and Klrg1 in each DC subset.(D)Gene Ontology(GO)enrichment terms of NK subsets(left:GO enrichment terms of NK-3; right: GO enrichment terms of NK-4). APAP-Rs: APAP-resistant group.
3.6. Macrophage subpopulations with an anti-inflammatory M2 functional phenotype are responsible for the tolerance to APAP hepatotoxicity
Macrophages are highly plastic and heterogeneous in liver injury [43]. In this study, macrophages were classified into four clusters, named Macro-1-4 (Fig. 6A). Compared with those in the APAP group,both the proportion and number of Macro-1 cells were decreased, while those of Macro-2-4 cells were increased in the APAP-resistant group (Fig. 6B). Macrophages are classified as proinflammatory M1 or anti-inflammatory M2 macrophages based on their cytokine types, cell surface markers, and transcriptional profiles [44]. The M1 and M2 phenotype scores identified Macro-1 cells as M1 macrophages (Fig. 6C). GO enrichment analysis showed that Macro-1 cells were involved in the regulation of leukocyte and granulocyte migration (Fig. 6D). Genes that recruit immune cells were highly expressed in Macro-1, including Thbs1,Cxcl2, and S100a8 (Fig. 6E). APAP increased the proportion and number of Macro-1 cells, thereby promoting inflammation and aggravating liver injury by enhancing the chemotaxis of inflammatory cells.In contrast,the M2 score was higher than the M1 score in Macro-2 and Macro-3 cells(Fig.6C).M2 macrophages have antiinflammatory effects, promote wound healing, tissue repair, and fibrosis, and are also involved in facilitating tumor growth and invasion [43]. GO enrichment analysis showed that Macro-3 cells were related to the positive regulation of the response to external stimuli and apoptotic cell clearance, which could resist APAPinduced liver injury (Fig. 6F). Macro-3 cells highly expressed Timd4 (Fig. 6E), which is responsible for preventing leukocyte transport,apoptotic cell recognition,and phagocytosis[45].During liver repair,M2 macrophages release anti-inflammatory cytokines,which are important in the remodeling of tissue, the resolution of inflammation,clearance of apoptotic bodies and tissue debris, and induction of angiogenesis [43]. Thus, Macro-3 cells may promote the repair of liver injury through the anti-inflammatory functions of M2 macrophages, contributing to autoprotection against APAP toxicity.Furthermore,Macro-2 cells regulated cholesterol and lipid biological functions and mediated tissue remodeling (Fig. 6G).Recent studies have shown that lipid metabolism is related to macrophage function,including their capacity for phagocytosis and cytokine secretion[46].In addition,Macro-2 cells highly expressed genes that promote tissue repair, such as Gpnmb and Igf1 [47,48].Thus, Macro-2 cells may alleviate APAP-induced liver injury via lipid metabolism and promotion of liver repair. No significant difference was found between the M1 and M2 scores in Macro-4 cells,which did not belong to any class of macrophages.

Fig.7. Neutrophil subpopulations with an N2 functional phenotype protect from acetaminophen(APAP)-induced liver injury.(A)The t-distributed stochastic neighbor embedding(t-SNE) plot of 6,185 neutrophils shows six clusters labeled as Neu-1-6. (B) Fraction of each neutrophil subset relative to all neutrophils (left) and the cell quantity of each subset(right).(C)A violin plot showing the N1 and N2 phenotype scores in each neutrophil subpopulation. ***P<0.001,ns:no significance.(D)Dot plot showing the expression of Camp and Ngp in each hepatocyte subpopulation. (E) Gene Ontology enrichment terms of Neu-6. APAP-Rs: APAP-resistant group; Neu: neutrophil.
CelltalkDB analysis showed that Macro-2 cells interacted with Endo-pc, Endo-pp, and LSECs through CCL4/atypical chemokine receptor 2 (ACKR2), CCL3/ACKR2, and CCL24/ACKR2 ligandreceptor pairs except for the Ccl2+subset (Fig. 6H). ACKR2 has a high affinity for pro-inflammatory cysteine-cysteine chemokines and promotes their intracellular degradation, thereby reducing local inflammatory levels [49]. ACKR2 plays an important role in limiting local inflammatory responses, inflammatory regression,and adaptive immune response regulation by clearing chemokines from tissues [50]. Therefore, Macro-2 cells inhibited inflammatory responses through cysteine-cysteine chemokines/ACKR2 ligandreceptor pairs, alleviating APAP-induced liver injury in the APAPresistant group.
3.7. Neutrophil subpopulations with an anti-inflammatory N2 phenotype protect from APAP-induced liver injury
We further analyzed the role of neutrophils in the adaptive response to APAP-induced liver injury. Neutrophils were classified into six clusters, named Neu-1-6 (Fig. 7A). Compared with those in the APAP group, the proportions of the Neu-3 and Neu-6 subsets increased, while those of Neu-2, -4, and -5 cells decreased in the APAP-resistant group (Fig. 7B). The N1 and N2 scores showed that Neu-1, -2, -4, and -5 cells were polarized into the N1 phenotype, while Neu-3 was polarized into the N2 phenotype(Fig. 7C). N1 polarized neutrophils have a pro-inflammatory phenotype [51]. Thus, when APAP-induced liver injury occurs,the proportion of Neu-2, -4, and -5 increases, thereby promoting inflammation and aggravating liver injury. In contrast, N2 neutrophils are associated with immune inhibition [52], which is consistent with the increase in Neu-3 levels in the APAP-resistant group. Compared with that in the APAP group, the N1/N2 ratio decreased in the APAP-resistant group (Fig. 7B). A decrease in the N1/N2 ratio is related to the resolution of inflammation [53]. In addition, the genes specifically expressed in Neu-3, such as cathelicidin antimicrobial peptide (Camp) and neutrophilic granule protein (Ngp),also suggest a role in promoting tissue regeneration(Fig. 7D). Camp is highly expressed in neutrophils from the center of the re-epithelialized wound bed during skin regeneration,indicating its ability to promote regeneration[54].In acid-induced acute lung injury, neutrophils promote the regeneration of alveolar epithelial cells by enhancing type II lung cell proliferation.Ngp is highly expressed in neutrophils and may be involved in this process [55]. The expression of Camp and Ngp in neutrophils increases during bone regeneration following maxillary sinus floor lifting[56].Therefore,Neu-3 cells may protect the liver from APAP hepatotoxicity by inhibiting inflammation and promoting regeneration. GO enrichment analysis showed that Neu-6 was closely related to ribosomal biological processes (Fig. 7E). Ribosome biogenesis is also an important mechanism of liver regeneration[57,58], thereby protecting the liver during adaptation to APAPinduced liver injury.
3.8. T cell subpopulations promote autoprotection against APAP by amplifying the IFN-γ response
T cells in the adaptation response to APAP-induced liver injury were classified into eight subgroups according to their specific gene expression(Fig.8A).Both the proportion and number of CD4+and CD8+naive T cell (Tn) subsets decreased in response to APAPinduced liver injury (Fig. 8B). CD4+Tn cells were related to T cell differentiation and activation(Fig.8C,left panel).CD8+Tn cells may mediate the regulation of neutrophil chemotaxis (Fig. 8C, right panel).They are activated upon antigen recognition,differentiating into central memory and effector T cells.Effector T cells can migrate to inflammatory sites to mediate the immune response [59]. In APAP-induced liver injury, the numbers of CD4+Tn and CD8+Tn cells may increase to support effector and memory T cells,resulting in severe liver damage. However, in the adaptation response to APAP-induced liver injury, the number of Tn cells decreases to reduce the inflammatory response.
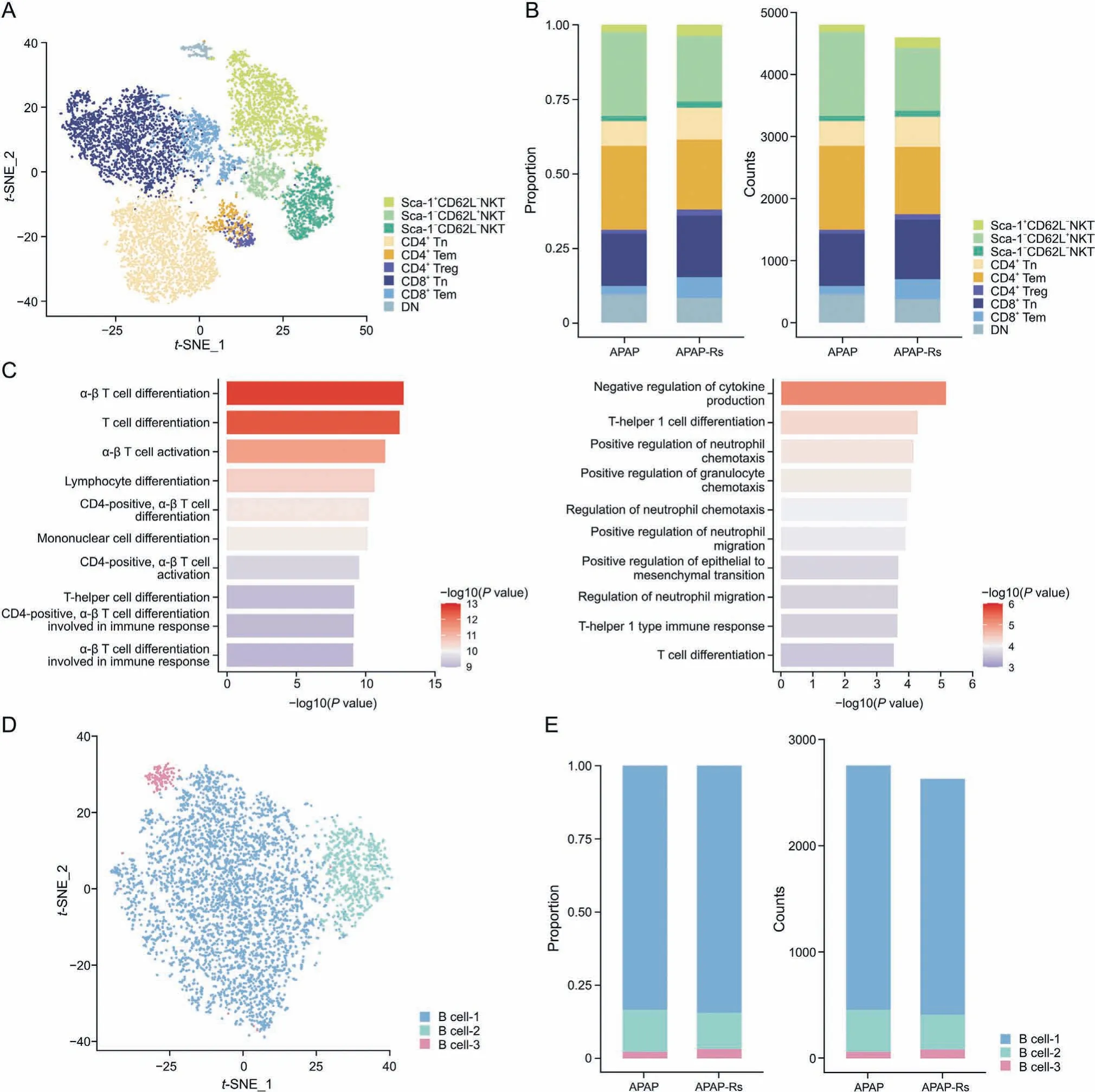
Fig. 8. T cell subpopulations promote autoprotection from acetaminophen (APAP). (A) The t-distributed stochastic neighbor embedding (t-SNE) plot of 9,404 T cells shows eight clusters labeled Sca-1+CD62L-natural killer T(NKT),Sca-1-CD62L+NKT,Sca-1-CD62L-NKT,CD4+naive T cell(Tn),CD4+Tem,CD4+Treg,CD8+Tn,CD8+Tem,and DN.(B)Fraction of each T cell subset relative to all T cells(left)and the cell quantity of each subset(right).(C)Gene Ontology(GO)enrichment terms of T cell subsets(left:GO enrichment terms of CD4+Tn;right:GO enrichment terms of CD8+Tn).(D)The t-SNE plot of 5,398 B cells shows three clusters labeled as B cell-1-3.(E)Fraction of each B cell subset relative to all B cells(left) and the cell quantity of each subset (right). APAP-Rs: APAP-resistant group.
In addition, compared with those in the APAP-induced liver injury group,the proportion and number of Sca-1-CD62L+natural killer T (NKT) cells in the resistant group were significantly increased(Fig.8B).Sca-1-CD62L+NKT cells may take up IL-12 and IL-2 and upregulate IL-18 receptor expression to amplify the IFN-γ response [60]. IFN-γ signaling plays an important role in liver regeneration and is driven by liver progenitor cells[61].At present,B cells in the adult mouse liver have not been mentioned in most reports[62];thus,B cells may play a less important role than other immune cells in the regulation of liver immune responses. In accordance with this,we did not observe a difference in the number and proportion of B cells between the APAP and APAP-resistant groups, suggesting that B cells may not be direct target cells (Figs.8D and E).
3.9. Dividing cells are increased in the APAP-resistant group
A total of 543 dividing cells were identified. This population of cells did not express hepatocyte-specific markers (Fig. S4) and did not belong to any NPC group. Compared with those in the APAP group,the proportion and number of dividing cells in the resistant group increased (Fig. 2C). GO enrichment analysis showed that dividing cells were closely related to DNA replication and chromosome division, indicating cell division ability (Fig. 9A), which was consistent with the results of the proliferation score (Fig. 9B).Therefore, the number of dividing cells was increased to promote the proliferation of liver cells, leading to liver tissue repair in response to APAP-induced liver injury.
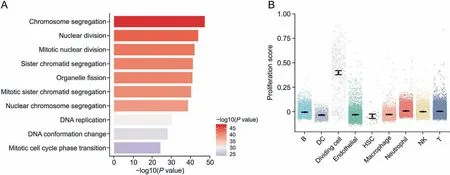
Fig. 9. Dividing cells are increased in the acetaminophen-resistant group. (A) Gene Ontology enrichment terms of dividing cells. (B) Proliferation scores for each type of nonparenchymal cells. DC: dendritic cell; HSC: hepatic stellate cell; NK: natural killer.
4. Discussion
Understanding the endogenous repair mechanisms of the autoprotective phenomenon in DILI is important for the prevention and management of this disease in clinical practice. However, to date,the underlying mechanisms have not been clearly elucidated because little is known about the dynamic regulation and biological functions of various liver cells involved in this process. ScRNA-seq offers the possibility of delineating the role and dynamics of different cells in the autoprotection from DILI. We used APAP as a model drug to investigate the transcriptional landscape of NPCs in a mouse model that is tolerant to APAP-induced liver injury, using single-cell RNA sequencing technology. Dynamic alterations of NPCs were identified, and the core gene expression signatures of key NPC subpopulations, as well as their interactions, were revealed during the development of tolerance to APAP hepatotoxicity.
Mice were pretreated with a mildly hepatotoxic dose of APAP,which resulted in protection from APAP hepatotoxicity upon subsequent dosing.The plasma concentration of APAP was observed to decrease after APAP autoprotection. Comparative analysis of endothelial cells from the livers of mice in the APAP and APAPresistant groups revealed obvious alterations in the subpopulations.It is worth noting that endothelial cells are important NPCs responsible for liver regeneration and resistance to liver injury. They can promote liver regeneration by increasing hepatocyte growth factor expression[63,64],and c-Kit-positive sinusoidal endothelial cells can enhance hepatocyte proliferation and alleviate CCl4-induced liver injury [65]. However, no studies have explored the role of endothelial cells in DILI resistance. In this study, we demonstrated that a Ccl2+subpopulation of endothelial cells is essential for APAP-induced liver injury. As an inflammatory chemokine, CCL2 plays a role in the migration and infiltration of immune cells, such as monocytes and macrophages [66,67]. The almost complete eradication of Ccl2+endothelial cells in the APAPresistant group indicated that inflammation was suppressed.Importantly, our functional study demonstrated that endothelial cells highly expressing Ccl2 can exacerbate hepatocyte injury after APAP exposure, implying that the inhibition of this subpopulation may alleviate APAP hepatotoxicity. The angiocrine factor RSPO3,which is highly expressed around the central vein[68],is a member of a novel secreted protein family that activates Wnt/β-catenin signaling in vertebrates and promotes the proliferation and angiogenesis of endothelial cells [69]. Endothelial Rspo3-driven WNT/Ca2+/Nuclear factor of activated T cells signaling is a key maintenance pathway for remodeling the vascular system [70],indicating that Rspo3+Endo-pc may support tolerance to APAPinduced liver injury by mediating angiogenesis through the RSPO3/Wnt signaling pathways. The effect of RSPO3 on tissue repair and homeostasis has also been demonstrated in intestinal lymphatic endothelial cells [71]. The results from our in vitro functional study are consistent with the theory that Rspo3+Endopc can promote hepatocyte proliferation after APAP exposure and suggest that this subpopulation is important in resistance to APAP hepatotoxicity. Further studies are needed to verify their potential contribution to anti-APAP hepatotoxicity and DILI in vivo.
DCs are a major determinant of intrahepatic immunity and undergo profound metabolic reprogramming in response to exogenous stimulation[72].We identified three subpopulations of pDCs in the liver and observed clear differences in their intracellular metabolism.Although detailed metabolic alterations of pDCs under physiological and pathological conditions have yet to be reported,this study detected a rapid induction of energy metabolismassociated pathways, including the glycolysis pathway, in DC-1 cells. The activation of glycolysis is critical for DC activation, and the rapid induction of this pathway in DCs allows them to respond quickly to dangerous signals caused by injury[72].Consistently,the increase in DC-1 cells may be a rapid response to toxic signals in the APAP group and may mediate APAP-induced liver injury through glycolysis and other energy metabolism pathways. A prominent feature of DC-2 and -3 cells is their marked expansion during the development of tolerance to APAP hepatotoxicity.DC-2 cells exhibit characteristics intermediate between those of DC-1 and DC-3 cells,while DC-3 cells display distinct intracellular metabolism compared to DC-1 cells. DC-3 cells specifically express mitochondrial genes and activate the oxidative phosphorylation pathway.The latter is closely associated with immature or tolerant DCs[73].Therefore, DC-3 cells constitute a group of drug-resistant DCs,resulting in low reactivity.Increased levels of DC-3 cells protect the liver from APAP-induced liver injury by inducing apoptosis or clearance of T cells,thereby suppressing the autoimmune response and the occurrence of inflammation [74]. In addition, DC-3 cells interacted with Endo-pc,Endo-pp,and LSECs through VEGFB/FLT1,VEGFA/PTPRB, and VEGFA/FLT1 ligand-receptor pairs, respectively.VEGFA stimulates many functions of endothelial cells, including proliferation and migration[75].The upregulation of VEGFR-1(also known as FLT1) and VEGFR-2 in LSECs facilitates their migration into hepatocyte clusters and enables the formation of capillaries[76,77]. Therefore, it is possible that DC-3 cells promote the angiogenesis of endothelial cells through VEGF-associated ligandreceptor pairs, thereby promoting liver tissue repair in the APAPresistant group. These findings provide new insights into the function of DCs in DILI and liver tissue repair.
A notable dynamic regulation of NK subpopulations is that they serve as mediators in the autoprotection from APAP hepatotoxicity through the IFN-γ pathway.Although it demonstrated that NK cells are involved in the pathogenesis of liver injury and fibrosis[78],NK cells producing IFN-γ have also been shown to repair liver diseases[39,40].In this study,NK-3 and NK-4 cells abundantly expressed KLR genes (which is associated with the expression of IFN-γ) [38] and may have alleviated liver damage through the IFN-γ pathway in the APAP-resistant group.However,the regenerative effects of NK cells depend on optimal NK cell activation.The overactivation of NK cells produces excess IFN-γ and leads to loss of self-tolerance, thereby impairing liver repair[79,80].Moreover,we observed that the levels of macrophages with an M2 phenotype (Macro-2 to Macro-4) and neutrophils with an N2 phenotype (Neu-3) significantly increased when developing tolerance to APAP hepatotoxicity, and the functions of these cells were related to the inhibition of inflammation and promotion of liver regeneration [43,52]. Additionally, Macro-2 cells interacted with Endo-pc,Endo-pp,and LSECs through CCL4/ACKR2, CCL3/ACKR2, and CCL24/ACKR2 ligand-receptor pairs,respectively. ACKR2 plays a role in the intracellular degradation of cysteine-cysteine chemokines,thereby reducing local inflammatory levels and alleviating APAP-induced liver injury[50].
5. Conclusion
Our results highlight that hepatic NPCs are the key determinants of resistance to APAP hepatotoxicity. The modulation of core signature genes such as Rspo3 and Ccl2 in endothelial cells and reprogramming of metabolic pathways in DCs may represent a novel strategy for the treatment of and defense against APAP toxicity and the promotion of liver regeneration.Although we have revealed evidence of interactions between several subpopulations,further investigation and functional verification are needed to identify the key intercellular communication networks among various NPCs,as well as between NPCs and liver parenchymal cells.This will help to develop effective strategies for DILI prevention.
CRediT author statement
Lingqi Yu:Methodology, Validation, Formal analysis, Investigation, Data curation, Writing-Original draft preparation, Writing-Reviewing and Editing, Visualization;Jun Yan:Methodology,Validation, Data curation, Writing-Original draft preparation,Visualization;Yingqi Zhan:Methodology, Validation, Data curation, Writing-Reviewing and Editing;Anyao Li:Methodology,Software,Data curation,Formal analysis;Lidan Zhu:Methodology,Validation, Data curation;Jingyang Qian:Methodology, Software,Data curation;Fanfan Zhou:Data curation,Project administration;Xiaoyan Lu:Conceptualization, Supervision, Project administration, Resources, Writing - Reviewing and Editing, Supervision,Funding acquisition;Xiaohui Fan:Conceptualization, Supervision,Project administration,Resources,Writing-Reviewing and Editing,Supervision, Funding acquisition.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (Grant No.: 81870426), the Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine(Grant No.:ZYYCXTD-D-202002),the Fundamental Research Funds for the Central Universities (Grant No.: 226-2023-00059), and the Fundamental Research Funds for the Central Universities.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2023.05.004.
 Journal of Pharmaceutical Analysis2023年8期
Journal of Pharmaceutical Analysis2023年8期
- Journal of Pharmaceutical Analysis的其它文章
- Integrative multi-omics and systems bioinformatics in translational neuroscience: A data mining perspective
- Ultrasensitive proteomics depicted an in-depth landscape for the very early stage of mouse maternal-to-zygotic transition☆
- Promise of spatially resolved omics for tumor research
- Temporal dynamics of microglia-astrocyte interaction in neuroprotective glial scar formation after intracerebral hemorrhage
- A single-cell landscape of triptolide-associated testicular toxicity in mice
- Single-cell and spatial heterogeneity landscapes of mature epicardial cells