
Single-cell and spatial heterogeneity landscapes of mature epicardial cells
2023-09-26 01:05:14JinlinDuXinYunHijunDengRongzhongHungBinLiuTinhuXiongXinglinLongLingZhngYingruiLiQingShe
Jinlin Du , Xin Yun , Hijun Deng , Rongzhong Hung , Bin Liu , Tinhu Xiong ,Xinglin Long , Ling Zhng , Yingrui Li ,**,1, Qing She ,*,1
a Department of Cardiology, The Second Affiliated Hospital of Chongqing Medical University, Chongqing, 400010, China
b Department of Nephrology, The Second Affiliated Hospital of Chongqing Medical University, Chongqing, 400010, China
c Key Laboratory of Molecular Biology for Infectious Diseases,Ministry of Education,Institute for Viral Hepatitis,The Second Affiliated Hospital of Chongqing Medical University, Chongqing, 400010, China
d Precision Medicine Center, The Second Affiliated Hospital of Chongqing Medical University, Chongqing, 400010, China
e Basic Medicine Research and Innovation Center for Novel Target and Therapeutic Intervention, Ministry of Education, The Second Affiliated Hospital of Chongqing Medical University, Chongqing, 400010, China
Keywords:
Epicardial cells
Gene markers
Single-cell sequencing
Spatial transcriptomics
A B S T R A C T
Tbx18, Wt1, and Tcf21 have been identified as epicardial markers during the early embryonic stage.However,the gene markers of mature epicardial cells remain unclear.Single-cell transcriptomic analysis was performed with the Seurat, Monocle, and CellphoneDB packages in R software with standard procedures. Spatial transcriptomics was performed on chilled Visium Tissue Optimization Slides(10x Genomics) and Visium Spatial Gene Expression Slides (10x Genomics). Spatial transcriptomics analysis was performed with Space Ranger software and R software.Immunofluorescence,whole-mount RNA in situ hybridization and X-gal staining were performed to validate the analysis results. Spatial transcriptomics analysis revealed distinct transcriptional profiles and functions between epicardial tissue and non-epicardial tissue. Several gene markers specific to postnatal epicardial tissue were identified,including Msln,C3,Efemp1,and Upk3b.Single-cell transcriptomic analysis revealed that cardiac cells from wildtype mouse hearts (from embryonic day 9.5 to postnatal day 9) could be categorized into six major cell types, which included epicardial cells. Throughout epicardial development, Wt1, Tbx18, and Upk3b were consistently expressed, whereas genes including Msln, C3, and Efemp1 exhibited increased expression during the mature stages of development. Pseudotime analysis further revealed two epicardial cell fates during maturation. Moreover, Upk3b, Msln, Efemp1, and C3 positive epicardial cells were enriched in extracellular matrix signaling.Our results suggested Upk3b,Efemp1,Msln,C3,and other genes were mature epicardium markers.Extracellular matrix signaling was found to play a critical role in the mature epicardium, thus suggesting potential therapeutic targets for heart regeneration in future clinical practice.
1. Introduction
Death due to cardiac ischemic injury remains among the most urgent problems worldwide [1]. The limited regeneration and repair ability of the human heart after ischemic injury has hindered efforts to increase the survival of patients with myocardial infarction (MI) [2]. The epicardium originates from proepicardial (PE)cells, serves as the source of progenitor cells during fetal development,and contributes to heart regeneration after injury[3,4].Adult epicardium has been suggested as a potential therapeutic target for heart regeneration in recent years [5,6]. Thymosin beta 4, a candidate for targeting the epicardium,has been found to improve cardiac functional repair after MI by increasing epicardial thickness and subepicardial vessel density [7]. Injection of vascular endothelial growth factor-A (VEGF-A) has been found to markedly improve cardiac function in a MI model of mouse via activation of adult epicardium [8]. However, gene markers of the mature epicardium remain unclear, thus impeding the understanding of the mechanisms of epicardium in cardiac repair.
Epicardial cells, a cluster of cells derived from proepicardium,play critical roles during cardiac development [3]. Epicardial progenitor cells give rise to epicardium-derived cells (EPDCs), and differentiate into several cardiac cell types, including coronary vascular smooth muscle cells [9], cardiac fibroblasts [10], endothelial cells [11], and adipocytes [12]. Tbx18, Wt1, and Tcf21 are commonly used as marker genes of epicardial cells;these cells have recently been found to display spatiotemporal transcriptional heterogeneity in different stages of heart development [13,14]. In heart development in chicks and mice, epicardial cells expressing Wt1 or Tbx18 overlap or are distinct from cells expressing Tcf21.These subsets of epicardial cells display differential regulation of cell differentiation and heart development [13]. Distinct subpopulations of epicardial cells with different functions have also been identified after MI [14].
Although early stage embryonic epicardial markers have been studied,specific markers of mature epicardial tissue have not been fully explored, thus preventing investigation of the functions and mechanisms of epicardial cells in cardiac repair after injury. Here,using the single-cell transcriptome data from public databases and spatial transcriptome data from our own assays, we revealed that
Mesothelin (Msln), epidermal growth factor (EGF)-containing fibulin extracellular matrix protein 1 (Efemp1), Uroplakin 3b (Upk3b), and Complement C3(C3),but not Tbx18,Wt1,or Tcf21,may be markers of mature epicardium.
2. Materials and methods
All procedures were approved by the Ethics Committee of Chongqing Medical University for the Ethics of Animal Experiments(Chongqing,China)(Approval No.:2022/231).Tbx18Cre mice were donated by Sylvia M. Evans [15]. R26RLacZ[16] Cre reporter mice were purchased from the Jackson Laboratory(Bar harbor,ME,USA).The methods of mouse genotyping and primer design were as previously described [15-17]. The Tbx18 probe plasmids were donated by Kispert and co-workers[18].Tbx18 expression analysis was performed on mouse embryos in whole-mount and sections through in situ hybridization, according to a previously described optimized method [19]. Homozygous Tbx18:Cre mice were identified by routine polymerase chain reaction. Double heterozygous Tbx18:Cre/R26RLacZmice were generated by crossing singleheterozygous mice. Immunofluorescence and X-gal staining experiments were performed on hearts to determine lineage specification, as previously described [20].
2.1. Spatial transcriptomics method
Heart tissue sections (embryonic days E10.5, E11.5, and E14.5 days,and postnatal days P1,P2,and P4)were placed on cold Visium Tissue Optimization Slides(10x Genomics)and Visium Spatial Gene Expression Slides (10x Genomics; Pleasanton, CA, USA). Subsequently, the samples were soaked in cold methanol and stained according to the Visium spatial gene expression(10x Genomics)or Visium spatial tissue optimization(10x Genomics)user guides.On the basis of tissue optimization time course investigations,18 min was chosen as the ideal time to permeabilize tissue for gene expression samples. According to the Visium Spatial Gene Expression user guide, a NovaSeq 6000 System (Illumina, San Diego, CA,USA) was used for loading (300 pM) and sequencing of libraries(approximately 250-400 M read-pairs for each sample and sequencing depth), together with a NovaSeq S4 Reagent Kit (200 cycles, 20,027,466, Illumina). The following read protocol was followed for the sequencing:read 1,28 cycles;i7 index read,10 cycles;i5 index read,10 cycles; read 2, 91 cycles.
2.2. Spatial transcriptomics data analysis
With Space Ranger software, raw FASTQ files and histology images were aligned with STAR v.2.5.1 b to the Cell Ranger hg38 reference genome “refdata-cellranger-GRCh38-3.0.0" (http://cf.10×genomics.com/supp/cell-exp/refdata-cellranger-GRCh38-3.0.0.tar.gz). The raw Unique Molecular Identifiers (UMI) count spot matrices, pictures, spot-image coordinates, and scale factors were imported to R. The spot matrix was filtered out to retain only the spots covering the tissue sections.The regularized negative binomial regression (SC transform) was used for normalization of raw UMI counts [21]. For dimensionality reduction, the top 2000 genes with the most variable expression levels were used. The Louvain algorithm (resolution = 0.5) was applied for clustering and clustering.The uniform manifold approximation and projection (UMAP) algorithms were used for visualization. A spot size scaling factor of 1.6 was used throughout the analysis for displaying cluster distributions spatially over hematoxylin and eosin images. The marker genes of one cluster were screened on the basis of statistically higher gene expression in the given cluster than in the remaining clusters according to the Wilcoxon rank-sum test,with a threshold of log2fold change(FC)>0.25,and expression in at least 20%of spots.
2.3. Bioinformatics analysis of single-cell RNA sequencing data
The processed single-cell RNA sequencing data(GSE193346)for the embryonic and postnatal heart tissues of two mouse strains(CD1 and C57BL/6)in 18 developmental stages were from study of Feng et al. [22]. The procedures of data processing were as previously described in detail [22]. Briefly, the reads were aligned and quantified in Cell Ranger software, sample demultiplexing was performed with the R package deMULTIplex, and multiple quality control metrics were calculated with the Scater R package. The Seurat R package[23]was used to create and merge Seurat objects and perform cell clustering. Batch correction was achieved with Harmony. Cell types were annotated carefully with a panel of lineage genes. With the processed and annotated data in RDS format, we conducted the downstream analysis with the Seurat R package. Marker genes of each cell type were identified with the FindAllMarkers function, with min. pct0.25 and logfc.threshold = 0.25 for both mouse strains.
Monocle [24] was used to conduct the pseudo-time analysis for epicardial cells. First, we constructed a CellDataSet object based on the downloaded Seurat object with the newCellDataSet function,then estimated size factors by with the estimateSizeFactors function and calculated dispersions with the estimateDispersions function.To choose genes defining progress (ordergenes), we used the highly variable genes identified by VariableFeatures function for C57BL/6.For CD1,we performed differential gene testing on expressed genes(defined as the genes expressed in at least ten cells) with the differentialGeneTest function with fullModelFormulaStr = ~stages. The top 2000 differentially expression genes (DEGs) were used as ordergenes.Dimensionality reduction of data was achieved with the reduceDimension function with the DDRTress method.The cells were ordered in pseudotime with orderCells with default parameters. To identify genes significantly correlated with pseudotime, we used the differential Gene Test function with parameter full Model For mula Str = ~sm.ns(Pseudotime). The BEAM function was used for the branched expression analysis modeling for branch point 2 in C57BL/6 and CD1. The enrichGO function in the clusterProfile R package[25]was used for Gene Ontology(GO)enrichment analysis.
CellphoneDB software (v4) [26] was used for cell communication analysis. The mouse gene IDs were converted to human homologs with the getLDS function in the R package biomaRt [27].CellphoneDB software was used for the analysis of cell-cell communication with the command “cellphonedb method statistical_analysis – counts-data hgnc_symbol – output-path./output –threshold 0.001 meta.txt count.txt,” and the dot plot and heat map were plotted with the commands “cellphonedb plot dot_plot” and “cellphonedb plot heatmap_plot”, respectively. The netVisual_circle function in the R package CellChat R package[28]was used for the network plot.The plot_cpdb function in the R package ktplots [29]was used to obtain dot plots with different gene families (gene.-family)including costimulatory,coinhibitory,chemokines,T-helper(Th) 1, Th2, Th17, Treg, and niche. The codes are provided in the Supplementary data.
2.4. Immunofluorescence staining
All immunofluorescence staining experiments were performed as previously described [30]. Tissues were soaked in 4% (V/V)paraformaldehyde for fixation, embedded, and then cut by cryosectioning to 10 μm. Nonspecific sites were blocked. Primary antibodies to Msln(ab168740;Abcam,Boston,MA,USA)were used.The sections were incubated with Alexa Fluor 594-conjugated secondary antibodies. Microscopy was performed with a laser scanning confocal microscope (Leica Microsystems GmbH, Heidelberg,Germany). The nuclei were stained with 4′,6-diamidino-2-phenylindole for 1-5 min.
2.5. X-gal staining
Whole-mount X-gal staining was performed as previously described[31].In brief,4%(V/V)paraformaldehyde(PFA)was used to fix samples for 15-60 min.Then 10%(V/V)Na deoxycholate and 10% (V/V) nonidet-P40 were used for permeabilization at room temperature. Samples were incubated in X-gal solution (50 mM potassium ferricyanide, 50 mM potassium ferrocyanide, 200 mM MgCl2,and 100 mg/mL X-gal in phosphate-buffered saline (PBS))for 4-12 h, then postfixed in 4% (V/V) PFA. For staining, sections were dehydrated via a graded sucrose solution series after being fixed in 4%(V/V)PFA.Subsequently,5-10 μm of cryosections were subjected to X-gal staining. Before staining, cryosections were refixed in 4% (V/V) PFA for 6-10 min.
2.6. Whole-mount RNA in situ hybridization
To visualize Tbx18 messenger RNA (mRNA), we performed whole-mount RNA in situ hybridization on mouse embryos as previously described through an optimized method[31].The probe plasmids of Tbx18 were constructed by Kispert and co-workers[32].For production of the labeled RNA probe, a digoxigenin RNA labeling kit and Dig detection kit (Roche, Basel, Switzerland) were used.
2.7. Statistical analysis
In spatial transcriptomic analysis, the marker genes of all clusters were analyzed using the Wilcoxon algorithm. The marker genes were then scored using the “group one vs the rest” approach.Significant marker genes for each cluster were selected based on their high specificity of expression, with log2FC > 0.25, and expression in at least 25%of spots.The DEGs were determined with the wilcox. test function from the stats R package, with a cutoff criterion of FC > 1.5 and P-adj. < 0.05. In single-cell sequencing analysis, marker gene of each cell types was identified by Findall-Markers functions in Seurat R packages,which used Wilcoxon Rank Sum test. DEGs between the cell type of interest and all other cell types were defined as genes with P value<0.01,log2FC>0.25,and only DEGs detected in a minimum fraction of 25%cells in either of the two populations were identified as marker genes.The enrichGO function in clusterProfile R package was used for GO enrichment analysis of genes, where P values were adjusted by false discovery rate, and cutoff of P value and q value were 0.05 and 0.2,respectively.
3. Results
3.1. Overview of the spatial landscapes of embryonic mouse hearts
The heart consists of three main components:the endocardium,myocardium, and epicardium. The embryonic epicardial tissue(EET) comprises a pool of progenitor cells with the potential to differentiate into multiple cell types and exhibits substantial histological heterogeneity with respect to the non-embryonic epicardial tissue (NEET) [33]. We used a combination of 10x Genomics spatial transcriptome technology and single-cell sequencing to identify distinct transcriptomic profiles of cardiac cells during mouse heart development (Fig. 1A). We specifically selected EET and NEET spots at E10.5, E11.5, and E14.5 on our spatial transcriptomic slices (Fig. 1B). A total of 1206 DEGs were identified between EET and NEET, and genes with the most significant changes in expression are displayed in Fig. 1C and Table S1. The spatial localization information for the top DEGs and several classical gene markers of the epicardium were visualized(Figs.1D and S1A). The 12 identified EET-specific marker genes were Upk3b,Upk1b,Aldh1a2,Gas1,Krt7,Colec12, Gucy1a1,Sema3d,Tbx18,Apela,Efemp1, and Bnc1. Notably, Upk3b, Upk1b, Sema3d, Wt1, Tcf21, and Tbx18 were previously described epicardial marker genes[34-37],whereas Lrrn4, Pkhd1l1, Efemp1, Tmem151a, and Krt7 were identified as novel gene markers of EET.However,Msln and C3 exhibited minimal expression in the early stages of epicardial development,whereas other gene markers were found to be expressed during those stages (Figs. 1D and S1A). Furthermore, the epicardial markers Wt1, Tbx18, Tcf21, Anxa8, Bnc1, Gata5, Gpm6a, Krt19, Krt8,Msln, and Myrf were expressed not only in EET but also in NEET(Figs. 1D and S1A). Functional enrichment analysis (Kyoto Encyclopedia of Genes and Genomes (KEGG) and gene set enrichment analysis(GSEA))of the DEGs revealed their significant enrichment in pathways associated with focal adhesion, the Wnt signaling pathway, and the mitogen-activated protein kinases (MAPK)signaling pathway (Figs. 1E and S1B). GO enrichment analysis indicated that the DEGs were enriched in processes such as epithelial tube morphogenesis, heart contraction, positive regulation of epithelial cell proliferation, heart development, epithelial cell proliferation, and the Wnt signaling pathway (Fig. 1F). These findings shed light on the DEGs and their functions between EET and NEET. Additionally, several novel gene markers of EET,including Lrrn4, Pkhd1l1, Efemp1, Tmem151a, and Krt7, were identified.

Fig. 1. Overview of the spatial landscapes of embryonic mouse hearts. (A) Flowchart of the study. V: ventricle; A: atrium. (B) Selected embryonic epicardial tissue (EET), nonembryonic epicardial tissue (NEET), postnatal epicardial tissue (PET) and non-postnatal epicardial tissue (NPET) spots on spatial transcriptomic slices. (C) Heat map of differential expression genes (DEGs) between EET and NEET. (D) Spatial transcriptome of epicardial gene markers at days E10.5, E11.5, and E14.5. (E) Kyoto Encyclopedia of Genes and Genomes(KEGG)and(F)Gene Ontology(GO)enrichment analyses of DEGs between EET and NEET.ScRNA-seq:single-cell RNA sequencing;DAPI:4',6-diamidino-2-phenylindole;MAPK: mitogen-activated protein kinases; GnRH: gonadotrophin-releasing hormone; VEGF: vascular endothelial growth factor.
3.2. Overview of the spatial landscapes of postnatal mouse hearts
Next, we conducted a comparison of the spatial transcriptional information between postnatal epicardial tissue (PET) and nonpostnatal epicardial tissue (NPET) from mouse heart tissue at P0 to P2. PET and NPET spots were selected on our spatial transcriptomic slices.A total of 1296 DEGs were identified,and the top significant DEGs are displayed in Fig. 2A and Table S2. We also visualized the spatial localization information for the top DEGs(Figs.2B and S2).Msln,Upk3b,C3,Efemp1,Lrrn4,Rspo1,Upk1b,Wt1,Tbx18, Tcf21, Anxa8, Bnc1, Gata5, Gpm6a, Krt19, Krt8, Myrf, Pkhd1l1,Tmem151a,and Nnat showed high expression in PET.Among them,Msln, Upk3b, Upk1b, Wt1, Tbx18, Tcf21, Anxa8, Bnc1, Gata5, Gpm6a,Krt19, Krt8, and C3 were recognized epicardial marker genes[34,37-39], whereas Efemp1, Lrrn4, Rspo1, Pkhd1l1, Tmem151a,Nnat, and Myrf were identified as novel gene markers of PET.Functional enrichment analyses(KEGG and GSEA)revealed that the DEGs between PET and NPET were enriched in the Wnt signaling pathway, cardiac muscle construction, extracellular matrix (ECM)-receptor interaction, and focal adhesion (Figs. 2C and S3A). GO enrichment analysis indicated that the DEGs were enriched in processes such as muscle cell differentiation, epithelial cell proliferation, regulation of the extracellular signal-regulated kinase 1(ERK1) and ERK2 cascade, cell-matrix adhesion, pericardium development,and negative regulation of transmembrane transport(Fig. 2D).
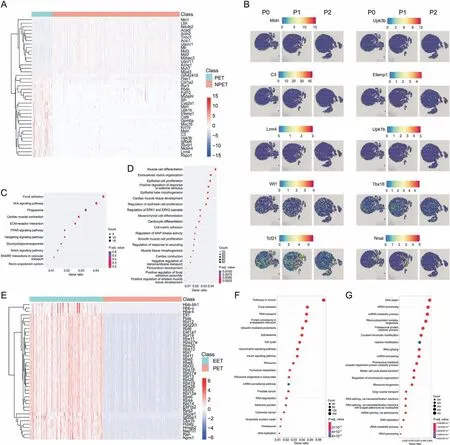
Fig. 2. Overview of the spatial landscapes of postnatal mouse hearts. (A) Heat map of differential expression genes (DEGs) between postnatal epicardial tissue (PET) and nonpostnatal epicardial tissue (NPET). (B) Spatial transcriptome of epicardial gene markers during days P0, P1, and P2. (C) Kyoto Encyclopedia of Genes and Genomes (KEGG) and(D) Gene Ontology (GO) enrichment analysis of DEGs between PET and NPET. (E) Heat map of DEGs between EET and PET. (F) GO and (G) KEGG enrichment analysis of DEGs between EET and PET.ECM:extracellular matrix;PPAR:peroxisome proliferator-activated receptor;SNARE:soluble N-ethylmaleimide-sensitive factor attachment protein receptor;ERK: extracellular signal-regulated kinase; MAP: mitogen-activated protein; mRNA: messenger RNA; ncRNA: noncoding RNA; rRNA: ribosomal RNA.
To identify the differential transcriptomic profiles between EET and PET, we further investigated the DEGs between these groups.Remarkably, the postnatal epicardium exhibited distinct transcriptional information from the embryonic epicardium.A total of 9585 DEGs were identified,and the top significant DEGs are displayed in Fig. 2E. Upk3b, Upk1b, Aldh1a2, Gas1, Sema3d, Wt1, Tcf21, Lrrn4,Pkhd1l1, Efemp1, Tmem151a, Krt7, Msln, C3, and Tbx18 showed differential expression between EET and PET (Fig. S3B). Functional enrichment analyses (KEGG and GSEA) revealed that the DEGs between PET and NPET were enriched in focal adhesion,ECM-receptor interaction,RNA transport,and soluble N-ethylmaleimide-sensitive factor attachment protein receptor(SNARE)interactions in vesicular transport(Figs.2F and S3C).GO enrichment analysis demonstrated that the DEGs were enriched in processes such as DNA repair,mRNA processing, ribonucleoprotein complex biogenesis, and covalent chromatin modification (Fig. 2G). These findings shed light on the DEGs and their functions between PET and NPET. Importantly,several novel gene markers of PET were identified, including C3,Efemp1, Lrrn4, Rspo1, Pkhd1l1, Tmem151a, and Nnat. Additionally,DEGs and their functions between EET and NEET were identified,thus indicating significant differences between EET and PET during development.
3.3. Embryonic and postnatal mouse epicardial cells show distinct transcriptional characteristics at the single cell level
The 10x Genomics spatial transcriptome of each Visium technology profiles spot had a diameter of 55 μm and captured approximately 1-15 cells. Therefore, we used single-cell sequencing analysis to explore the epicardium transcriptional information at the single cell level (GSE193346). This single-cell dataset included cardiac cells from wildtype mouse hearts (CD1 and C57BL/6)at 18 developmental stages(from embryonic day 9.5 to postnatal day 9), as profiled with the 10x Genomics single-cell RNA-seq platform. Cell clusters were visualized through UMAP plots.The cell types were annotated with a panel of lineage genes[22].A total of 27,445 and 25,436 cells in the dataset from CD1 and C57BL/6 mice, respectively, were used for analysis. The cells were classified into six major cell types in CD1 and C57BL/6 mice: cardiomyocytes,endothelial cells,epicardial cells,fibroblast-like cells,immune cells, and blood cells (Figs. 3A and S4A). The gene list of markers of every cell type is displayed in Tables S3 (for CD1 mice)and S4(for C57BL/6 mice).The top ten marker genes for epicardial cells in CD1 mice included Upk3b, C3, Msln, Itm2a, Igfbp6, S100a6,Krt19,Sulf1,Mgst1,and Rarres2(Fig.S5A).In addition,the following markers were specifically expressed in the epicardium in CD1 mice:Wt1, Tcf21, C3, Tbx18, Upk1b, Upk3b, Msln, Bnc1, Gpm6a, Krt8, Myrf,Lrrn4, Pkhd1l1, Efemp1, Tmem151a, Gata5, Krt19, Nnat, and Rspo1(Figs. 3B, 3C, and S6A). However, Wt1, Tcf21, Tbx18, Gpm6a, Krt8,Gata5, and Nnat were also expressed in non-epicardial cells.Furthermore,violin plots revealed the expression patterns of these epicardial markers: Wt1, Tbx18, Upk1b, and Upk3b were expressed in all stages during epicardial development;the expression levels of Msln, C3, Efemp1, Gpm6a, Tmem151a, Rspo1, Lrrn4, and Pkhd1l1 gradually increased during development; and the expression of Tcf21 and Nnat subsequently decreased in mature epicardium(Fig. 3D). Furthermore, the localization of Tbx18 expression was examined with in situ hybridization and X-gal staining, both of which suggested that Tbx18 was a marker of proepicardium cell and epicardial cell clusters during early stages of heart development(Fig. 3E). Furthermore, C57BL/6 mice also showed a similar expression pattern (Figs. S4B-D,S5B, and S6B).

Fig. 3. Gene expression patterns of epicardial cells in CD1 mice during development. (A) Uniform manifold approximation and projection (UMAP) of cardiac cells during development (from embryonic day 9.5 to postnatal day 9). (B) Dot plot of gene expression patterns in the epicardium. (C) Feature plot on UMAP of epicardial gene markers in the epicardium during development.(D)Violin plots of epicardial gene markers in distinct stages during development.(E)Tbx18 messenger RNA(mRNA)and Lacz expression at E10.5,on the basis of in situ hybridization (ISH) and X-gal staining, respectively. V: ventricle; A: atrium; ProEPCs: proepicardium cells; EPCs: epicardium cells.
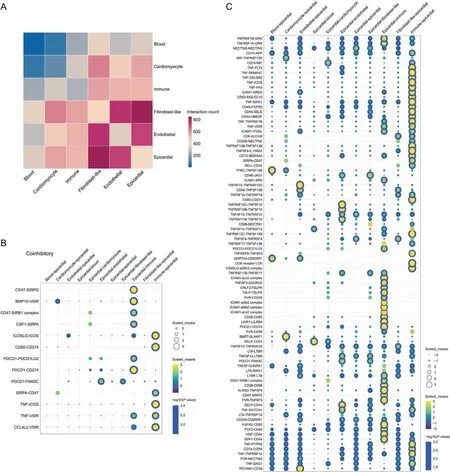
Fig.4. Communication between epicardial cells and other cardiac cells in CD1 mice during development.(A)Heat map of interactions between epicardial cells and other cardiac cell types during development. (B) Dot plot of the coinhibitory network between epicardial cells and other cardiac cell types during development. (C) Dot plot of the costimulatory network between epicardial cells and other cardiac cell types during development. SIRPG: signal regulatory protein gamma; BMP: bone morphogenetic protein; VSIR: V-set immunoregulatory receptor; CSF1: colony stimulating factor 1; SIRPA: signal regulatory protein alpha; ICOS: inducible T cell costimulatory; PDCD1: programmed cell death 1;FAM3C:FAM3 metabolism regulating signaling molecule C;TNF:tumor necrosis factor;CCL4L2:C-C motif chemokine ligand 4 like 2;TNFRSF:TNF receptor superfamily member;GRN: granulin precursor; NECTIN: nectin cell adhesion molecule; APP: amyloid beta precursor protein; MIF: macrophage migration inhibitory factor; FLT: fms related receptor tyrosine kinase; SEMA: semaphoring; CELSR: cadherin EGF LAG seven-pass G-type receptor; FAS: Fas cell surface death receptor; ICAM: intercellular adhesion molecule; AREG:amphiregulin; SIGLEC: sialic acid binding Ig like lectin; RIPK1: receptor interacting serine/threonine kinase; FGFR: fibroblast growth factor receptor; SELE: selectin E; HBEGF:heparin binding EGF like growth factor;ITGAL:integrin subunit alpha L;ALCAM:activated leukocyte cell adhesion molecule;TNFSF:TNF superfamily member;IL13RA2:interleukin 13 receptor subunit alpha 2; SELL: selectin L; TFRC: transferrin receptor; JAG: jagged canonical Notch ligand; SPN: sialophorin; ADIPOQ: adiponectin, C1Q, and collagen domain containing; LCK: LCK proto-oncogene; ADGRG: adhesion G protein-coupled receptor; CRLF: cytokine receptor like factor; TSLP: thymic stromal lymphopoietin; TSLPR: TSLP receptor; PVR: PVR cell adhesion molecule; LAIR1: leukocyte associated immunoglobulin like receptor 1; LILRB4: leukocyte immunoglobulin like receptor B4; SLAMF1: signaling lymphocytic activation molecule family member 1;SELP:selectin P;LTA:lymphotoxin alpha;LTBR:lymphotoxin beta(LTB)receptor;KLRG2:killer cell lectin like receptor G2;FGF:fibroblast growth factor; HGF: hepatocyte growth factor; SPP: secreted phosphoprotein;PTPRS: protein tyrosine phosphatase receptor type S; COPA: coat complex subunit alpha;DAG: dystroglycan; PECAM: platelet/endothelial cell adhesion molecule.
To obtain spatial information for the epicardial cells, we obtained the overlapping marker genes among epicardial cells from CD1, C57BL/6, and EET (DEGs between EET and NEET with FC values > 2), and identified 68 overlapping genes (Fig. S6C and Table S5), including the known epicardial markers Tbx18, Upk1b,Upk3b, Msln, Bnc1, Gpm6a, Krt8, Myrf, and several novel epicardial markers including Lrrn4, Pkhd1l1, Efemp1, and Tmem151a. The overlapping genes among epicardial cells from CD1, C57BL/6, and PET (DEGs between PET and NPET with FC values > 2) were obtained, and 69 overlapping genes were identified (Fig. S6D and Table S6), including the known epicardial markers Msln, Upk3b,Upk1b, Wt1, Gpm6a, Krt19, Krt8, Myrf, and Nnat, and the novel epicardial markers C3, Efemp1, Lrrn4, Rspo1, and Pkhd1l1. These results revealed the temporal heterogeneity of hub genes of the epicardium during development at the single-cell level. The observed increase in expression of Msln, C3, Efemp1, Rspo1, and Lrrn4 in mature epicardium indicated that these genes may play crucial roles in mature epicardium.
3.4. Cell communication between epicardial cells and other cardiac cells during development
To further explore the interactions between the epicardial cell cluster and non-epicardial cell clusters at the single-cell level, we conducted cell communication analysis (Fig. 4). Epicardial cells interact with various cell types (endothelial cells, fibroblast-like cells, immune cells, blood cells, and cardiomyocytes) during development through ligand-receptor interactions. For example,they interact with immune cells via TNF-FLT4,TNF-RIPK1,or CD44-FGFR2, with cardiomyocytes through BMP7-SLAMF1, BMP10-VSIR,or CD74-MIF,and with fibroblast-like cells through CD46-JAG1 and PDCD1-FAM3C (Figs. 4B and C). C57BL/6 mice also displayed various ligand-receptor interactions between epicardial cells and non-epicardial cells (Fig. S7). These results indicated the ligandreceptor interactions between epicardial cells and other cell types during development, thus suggesting that epicardial cells are involved in an intricate network of signaling pathways with other cells during development.
3.5. Temporal heterogeneity trajectories of epicardial cells during development
The gene expression patterns of epicardium during development showed temporal heterogeneity, and significant differences were observed in gene expression and enrichment functions between EET and PET. Therefore, further exploration of the developmental trajectory of epicardial cells is critical for comprehensive understanding of their physiological functions. Pseudo-time analysis conducted for epicardial cells revealed the three branches and seven states in the development trajectory (Figs. 5A and S8A).Along the developmental trajectory, the expression of Msln, C3,Efemp1,and Gpm6a gradually increased,whereas the expression of Tcf21 and Nnat decreased. Upk3b, Upk1b, Wt1, and Tbx18 showed consecutive expression during epicardium development (Figs. 5B and S8B).Two fates for epicardial cells were observed during their maturation in CD1 mice(Fig.5A).To reveal the differences between these fates, we conducted GO enrichment analysis. Genes in cell fate 1(the upper branch)were enriched in upregulation of reactive oxygen species metabolic process and oxygen transport, and downregulation of cytoplasmic translation, ribosome biogenesis,and regulation of cell-substrate junction assembly (Fig. 5C). Genes in cell fate 2(the lower right branch)were enriched in upregulation of angiogenesis, regulation of vasculature development and necrotic cell death, downregulation of DNA methylation or demethylation, and multicellular organism growth. C57BL/6 mice also exhibited similar temporal expression patterns (Figs. S9 and S10).These results revealed the differential expression of genes during epicardial development at the single-cell level, thus highlighting significant changes in gene expression during development. Our findings suggest that mature epicardial cells may display distinct gene expression patterns and functions from those of the embryonic epicardium. Furthermore, epicardial cells differentiated into two distinct cell fates with different functions during the mature stage,thus indicating that mature epicardial cells may have specific and distinct roles in heart development and maintenance.
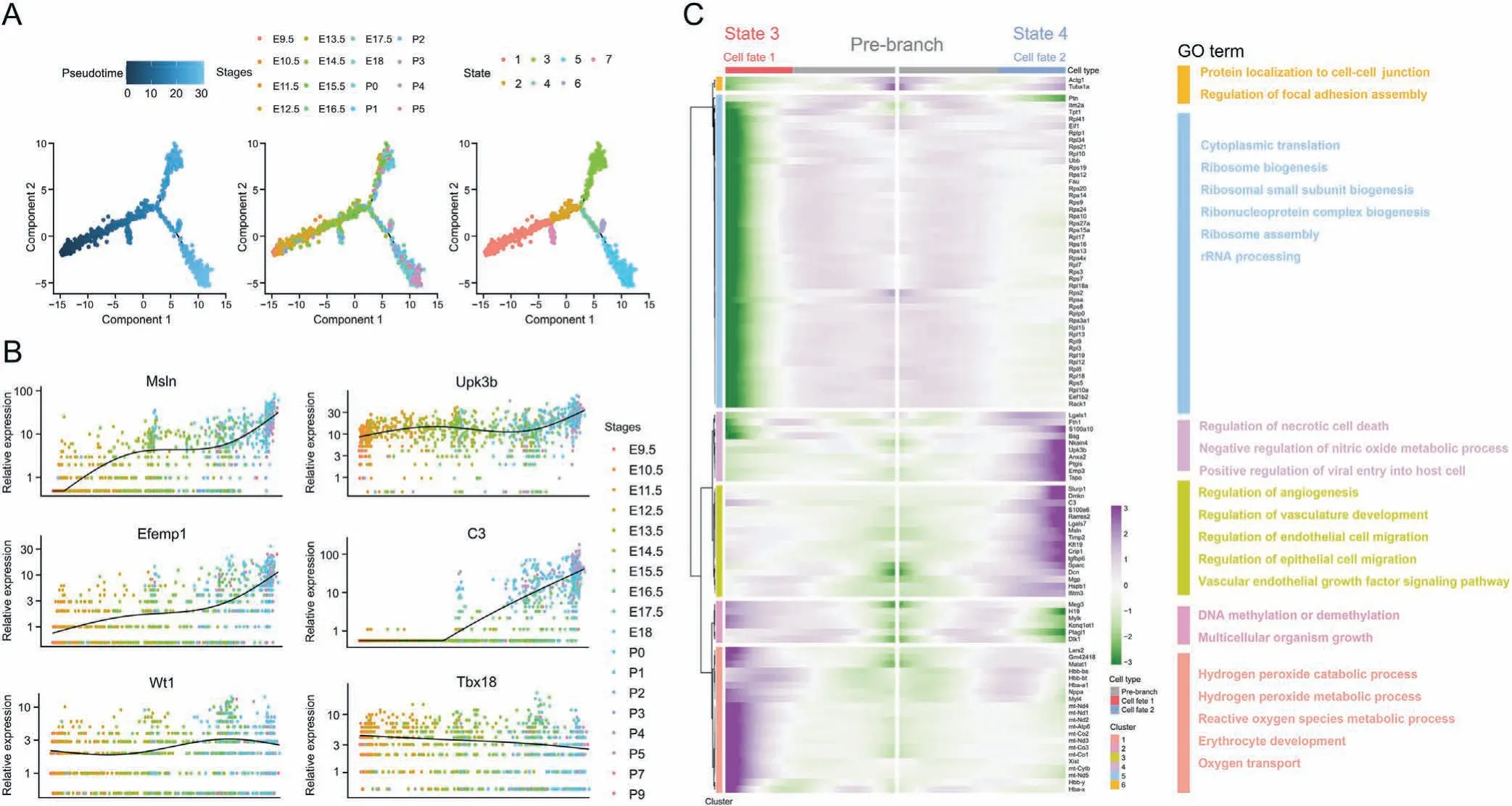
Fig. 5. Temporal heterogeneity trajectories of epicardial cells in CD1 mice during development. (A) Pseudotime analysis of epicardial cells during development. (B) Jitter plot of epicardial gene markers during development. (C) Heat map and Gene Ontology (GO) analysis of two fates for epicardial cells during their maturation. rRNA: ribosomal RNA.
3.6. The regulatory network of Msln, Upk3b, C3, and Efemp1 positive cells in mature epicardium
To investigate the functions of Msln and Efemp1 positive cells in the mature epicardium, we selected the Msln and Efemp1 positive cells from PET and NPET spots in our spatial transcriptomic slices.A total of 748 DEGs were identified between PET-Msln and NPET-Msln positive cells.The top significant DEGs,including C3,Upk3b,Col1a1,Msln, Rarres2, Col1a2, Efemp1, Gpx3, Gm42418, and Igfbp6, are displayed in Fig. 6A and Table S7. KEGG analysis revealed that these DEGs were enriched in pathways such as axon guidance, focal adhesion,the Wnt signaling pathway,and the transforming growth factor (TGF)-beta signaling pathway. GO enrichment analysis showed that the DEGs were enriched in processes such as ECM organization, extracellular structure organization, and epithelial cell proliferation (Fig. 6B). For PET-Efemp1 and NPET-Efemp1 positive cells,the top ten significant DEGs were Irx5,Msln,C3,Upk3b,Rarres2,Gm42418, Irx2, Sftpa1, Gpc3, and Nr0b2 (Fig. 6C and Table S8).Functional enrichment analysis also indicated that these DEGs were enriched in focal adhesion, ECM organization, extracellular structure organization,and collagen metabolic process(Fig.6D).
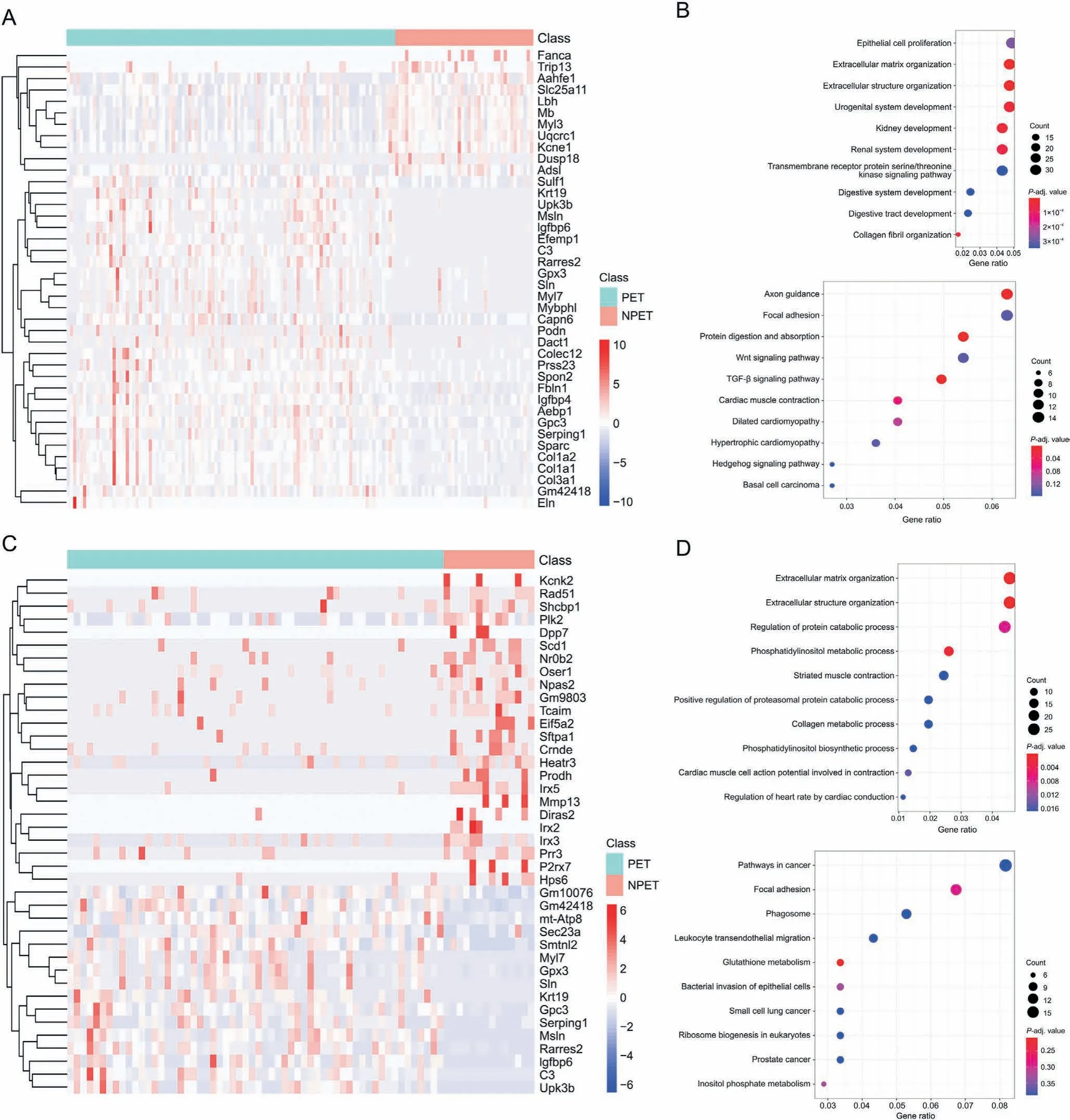
Fig.6. Differential transcriptional profiles and functions between postnatal epicardial tissue(PET)and non-postnatal epicardial tissue(NPET)in Msln+and Efemp1+cells.(A)Heat map of differential expression genes(DEGs)between PET-Msln+and NPET-Msln +cells.(B)Gene Ontology(GO)and Kyoto Encyclopedia of Genes and Genomes(KEGG)enrichment analysis of DEGs between PET-Msln+ and NPET-Msln + cells. (C) Heat map of DEGs between PET-Efemp1+ and NPET-Efemp1+ cells. (D) GO and KEGG enrichment analysis of DEGs between PET-Efemp1+ and NPET-Efemp1+ cells. TGF: transforming growth factor.
Next,the transcriptional regulatory signaling networks of Msln,Upk3b, and C3 positive mature epicardial cells were explored through enrichment analysis.We defined count values>0 for Msln,Upk3b,and C3 as positive cells.Furthermore,these positive cells or spots in spatial transcriptomic data were subjected to further functional enrichment analysis. The GO and KEGG enrichment analyses indicated similar results for Msln, Upk3b, and C3 positive cells (Figs. 7A-F). Interestingly, focal adhesion and ECM-receptor interaction were all involved in the regulatory network of Msln,Upk3b, C3 positive cells, thereby indicating the major role of ECM signaling in mature epicardium. The focal adhesion function mediated by ECM signal clearly played an important role in the regulation signal of Msln,Upk3b,and C3 positive epicardial matrix.The protein-protein interaction network of KEGG functions of focal adhesion was determined with STRING database, which revealed that genes including Arhgap5,Gsk3b,Itga5,Pgf,Pdgfrb,Rock2,Actn3,Parvb, Parvg, and Mapk1 in focal adhesion signaling, and Itga5,Lama2, Dag1, Agrn, Itga8, and Col6a6 in ECM-receptor interaction signaling, were involved in its signal regulation (Fig. 7G). Importantly, the expression of the specific markers of Msln+in mature epicardium was validated by immunostaining of mouse heart at P0 and P2 days(Fig.7H).The results revealed that ECM signaling may play a crucial role in Msln, Upk3b, C3, and Efemp1 positive cells in the mature epicardium.
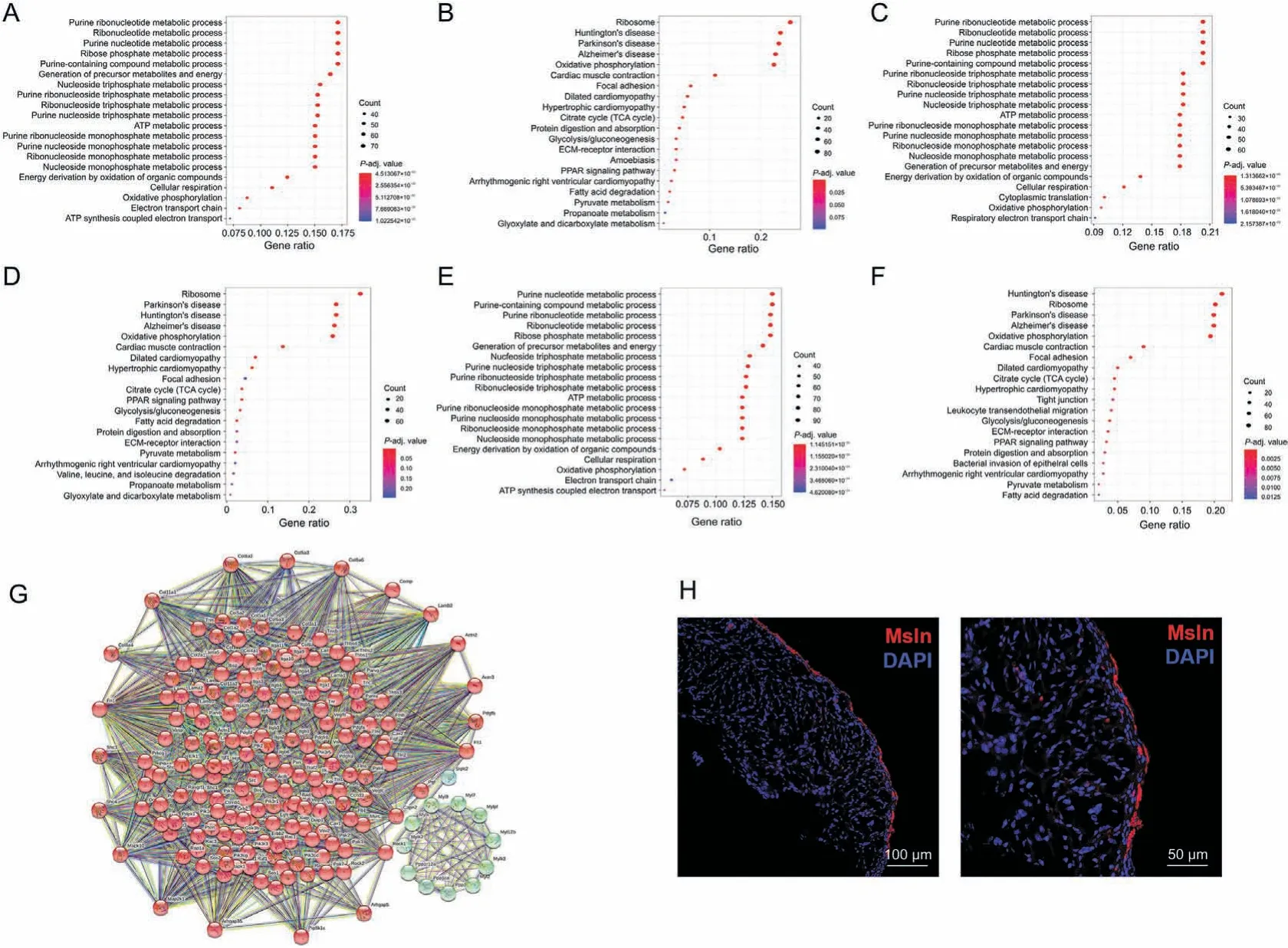
Fig. 7. Regulatory network of Msln, Upk3b, and C3 positive cells in mature epicardium. (A) Gene Ontology (GO) enrichment analysis of Msln positive cells on the basis of spatial transcriptomic data.(B)Kyoto Encyclopedia of Genes and Genomes(KEGG)enrichment analysis of Msln positive cells,on the basis of spatial transcriptomic data.(C)GO enrichment analysis of Upk3b positive cells,on the basis of spatial transcriptomic data.(D)KEGG enrichment analysis of Upk3b positive cells,on the basis of spatial transcriptomic data.(E)GO enrichment analysis of C3 positive cells,on the basis of spatial transcriptomic data.(F)KEGG enrichment analysis of C3 positive cells,on the basis of spatial transcriptomic data.(G)Protein-protein interaction network analysis of KEGG functions of focal adhesion in Msln + cells. (H) Expression of Msln, detected by immunofluorescence staining in the mouse heart during postnatal days. ATP: adenosine triphosphate; TCA: tricarboxylic acid; ECM: extracellular matrix; PPAR: peroxisome proliferator-activated receptor; DAPI: 4′,6-diamidino-2-phenylindole.
4. Discussion
Single-cell sequencing and spatial transcriptomics have been widely applied worldwide because of their unique advantages.Single-cell sequencing enables exploration of the transcriptomes of individual cells and subsets of cells of interest. Integration with spatial transcriptomics can identify the locations of given gene sets in specific cell subpopulations [40]. Here, we determined the single-cell and spatial landscapes of epicardial cells in mouse heart tissue from embryonic day 9.5 to postnatal day 9. Cellular and temporal heterogeneity was found in epicardial cells, and distinct developmental stages of epicardial cells displayed different gene markers and cellular functions. Further analysis indicated that Upk3b was a gene marker of epicardial cells during early embryonic heart development. Upk3b, Msln, Efemp1, and C3 were also identified as markers of mature epicardium. Functional analysis showed that ECM signaling plays a major role in the mature epicardium.
4.1. Cellular and temporal heterogeneity of epicardial cells
PE is a conserved transient population of ancestral cells. In mouse embryos, cells derived from the PE undergo migration events around embryonic day 9.5[41].These PE-derived cells form a monolayer of mesothelial cells known as the epicardium, which lines the outermost layer of the heart. Initially, the epicardium exhibits epithelial-like characteristics and acts as a barrier between the primitive myocardium and the pericardial cavity. However, a subset of epicardial cells undergo detachment from the epicardium and invade the myocardium,thus giving rise to EPDCs around E12.5[42]. Subsequently, the EPDCs undergo a complex process called epithelial-to-mesenchymal transition (EMT), thus leading to their differentiation into various cardiac cell lineages. Several genes,including Wt1, Tbx18, Tcf21, Sema3d, Sox9, Aldh1a1, Upk3b, and Upk1b, exhibited significant changes in expression during the proepicardium stage.Some of these genes showed peak expression at E11.5 and subsequent attenuation [37,43,44]. By integrating single-cell sequencing and spatial transcriptome technology, we identified distinct expression patterns of epicardial gene markers.Some markers, such as Tbx18, Wt1, Upk1b, and Upk3b, maintained consistent expression in the epicardium from EET to PET, in agreement with findings from recent studies [37]. However, the expression of certain genes,such as Msln,C3,and Efemp1,increased around E12.5-E14.5. Meanwhile, some genes exhibited high expression in EET but not PET, such as Nnat and Tcf21, thus indicating the cellular and temporal heterogeneity of epicardial cells during development.
The epicardium has functional heterogeneity in heart development in chicks,mice,zebrafish,and epicardium derived from human pluripotent stem cells, and demonstrates distinct functions from those of non-cardiac tissues [13,45,46]. Our findings revealed DEGs and their spatial distribution between the epicardium and noncardiac epicardium tissues. These genes included classic epicardial gene markers, such as Tbx18, Wt1, and Tcf21, as well as novel epicardial gene markers, such as Msln, Efemp1, C3, Lrrn4, and Nnat.Furthermore, the DEGs were enriched in pathways associated with Wnt signaling,focal adhesion,and cell differentiation.Recent studies have suggested that Wnt signaling exhibits biphasic and antagonistic effects on cardiac specification and differentiation,depending on the stage of embryogenesis [47]. The activation and inhibition of Wnt signaling in human pluripotent stem cells in different stages influences cardiomyocyte differentiation [48]. Our results also indicated distinct enrichment pathways between the epicardium and non-epicardium,thus highlighting the different functions of epicardial cells during development. Additionally, analysis of cell communication revealed that epicardial cells interact with various cell types, including endothelial cells, fibroblast-like cells, and cardiomyocytes, through ligand-receptor interactions. The results suggested that TNF-FLT4, BMP7-SLAMF1, FGF2-CD44, CD44-HBEGF,and other interactions may play major roles in the crosstalk between epicardial cells and other cell types. However, the network of signaling pathways involved in development is intricate. These findings may provide novel targets for future research aimed at elucidating the roles of epicardial cells in development.
Temporal heterogeneity was also observed in epicardial cells,including distinct transcriptional differences at various stages from embryonic day 9.5 to postnatal day 9.Genes such as Tbx18,Wt1,Tcf21,Upk1b, and Upk3b exhibited continuous expression in both embryonic and mature epicardial cells.Interestingly,Tbx18,Wt1,and Tcf21 were expressed not only in the epicardium but also in the myocardium,thus suggesting their migration into the myocardium through EMT [44]. During development, certain epicardial gene markers showed increased expression and maintained their expression in the mature epicardium. These markers included Msln, Efemp1,Tmem151a,Lrrn4,Rspo1,and C3.In contrast,the expression of other epicardial gene markers decreased during development, such as Tcf21 and Nnat.The period between E12.5 and E14.5 is crucial for the differentiation of epicardial cells.Some epicardial cells undergo EMT and differentiate into other cell types,whereas others remain in the epicardium [37]. This temporal heterogeneity in gene expression may be attributable to these dynamic processes occurring during epicardial development. Pseudotime analysis further indicated that epicardial cells in mature stage differentiated into two distinct cell fates: one cell fate may be involved in vascular development,whereas the other may be involved in hydrogen peroxide, reactive oxygen species metabolic process, and cardiac contraction. Additionally, cells in mature stages displayed enrichment in collagencontaining ECM. The ECM plays an important role in embryonic heart development,including guiding the maturation of cardiac cell types and providing environment for stem cells [49]. However,recent studies have suggested that the ECM may be involved in an important mechanism leading to the loss of pluripotency of progenitor cells and to the negative regulation of progenitor cell proliferation[50,51].Therefore,the epicardial cells in mature stages may play critical roles in heart development by shaping the microenvironment, and may also negatively regulate progenitor cells proliferation after cardiac injury. More studies are needed to clarify the complicated functions of mature epicardial cells.
4.2. Subpopulations with potential gene markers of mature epicardial tissue
Uroplakins (Upks) are a family of proteins, including two tetraspanins Upk1a and Upk1b, and three single transmembrane proteins Upk2, Upk3a, and Upk3b [52]. As the major protein component of conserved urothelium membrane,Upk3b combining with Upk1b participates the construction of mammalian urothelium [53]. Upks are commonly believed to be associated with diseases of the urinary system, such as urothelial cancer or urinary tract infections [54]. However, recent studies have indicated the important role of Upks in embryogenesis. Mice with disrupted expression of Upk1b show abnormal development of bladder urothelium and disruption of urothelial plaques in the bladder and kidneys [55]. Upk3b has been detected in neural crest cells, heart,and kidneys in mouse embryos,and has been found to be involved in the maturation of gametes and gamete-delivery organs [56].Single-cell sequencing and spatial transcriptomics analysis indicated that Upk3b,like Tbx18,Wt1,and Tcf21,was an early embryonic marker of (pro)epicardial cells during heart development. These cells were also enriched in ribosome metabolism, in line with recent studies indicating that ribosomes with the ability to regulate translation are essential for early embryogenesis and tissue development [57,58]. Furthermore, Upk3b was also highly expressed during P1-4, thus indicating its potential as a marker of mature epicardium.
Efemp1,a member of the fibulin family of proteins,is known for its involvement in ECM organization and stability.Previous studies have highlighted the role of Efemp1 in maintaining ocular tissues,such as the retina and the elastic fibers of the lens [59]. Efemp1 expression has also been observed in various adult tissues,including the heart, lung, and skeletal muscle [60]. During embryonic development,murine Efemp1 mRNA expression initiates at embryonic day 9.5 in condensing mesenchyme structures and persists throughout development, thus contributing to the formation of bone,cartilage,and various skeletal structures,including the craniofacial, appendicular, and axial skeleton [61]. Recent studies have elucidated the role of Efemp1 in the progression of myocardial fibrosis in diabetes-induced cardiomyopathy, as well as its association with the prognosis of patients with acute MI [62,63]. However, limited research has explored the relationship between Efemp1 and the epicardium.Our findings suggested that Efemp1 is a marker of mature epicardium.Additionally,GO and KEGG analyses indicated that Efemp1-positive epicardial cells were more enriched in ECM signaling than Efemp1-positive non-epicardial cells. These findings suggest that Efemp1 plays an important role in the mature epicardium by regulating ECM signaling pathways.
Msln, a protein anchored to the cell surface via glycosylphosphatidylinositol, plays a critical role in cell adhesion. This protein is overexpressed in epithelial mesotheliomas, ovarian cancers, and specific squamous cell carcinomas [64]. Diseases associated with Msln include benign mesothelioma and asbestosis.Previous research has revealed that Msln is specifically and highly expressed in cancer tissues [65]. Its related pathways include metabolism of proteins and cellular adhesion.Membrane-anchored forms may have roles in cellular adhesion [66]. Mesothelintargeted CAR-T cell therapy has achieved excellent therapeutic effects against cancer[66].However,few studies have examined the expression pattern and function of Msln in the heart.Single-cell and spatial transcriptomics analyses revealed that Msln was also a marker of mature epicardium. GO and KEGG analyses indicated enrichment in focal adhesion and ECM signaling in Msln positive epicardial cells. Protein-protein interaction network analysis highlighted several genes,including Vegfb,Fn1, Flt1, Mapk1,Pdgfrb,Src, and Rac, which may play roles in epicardial development through focal adhesion and ECM-receptor interaction signaling pathways. These findings are consistent with those from previous studies[67-70].Prior research has demonstrated the involvement of Src and Rac in the EMT of epicardial cells during embryogenesis[67,68]. Pdgfrb has been shown to play a role in coronary development, because the coronary vasculature differentiates primarily from epicardial cells through the EMT process [69]. Moreover, our previous study has indicated that the MAPK pathway is involved in thyroid hormone-mediated promotion of epicardial cell proliferation [70].
C3 is an indispensable part of the complement system. Its activation is necessary for classical, alternative and lectin pathways of complement activation. C3 is generated by the liver, activates macrophages and adipocytes in the liver, and plays an important role in the immune system[71].Recent studies have indicated that C3 mediated immune signaling is involved in early neural brain development. C3 disruption results in impairment of neuronal migration as well as neuronal stem cell proliferation and differentiation in the developing brain[72].However,the role of C3 in heart development remains unclear. The results also indicated that C3,like Upk3b and Msln, was a potential gene marker of mature epicardial tissue.
4.3. ECM signaling enrichment in Upk3b+, Efemp1+, Msln+, and C3+ epicardial cells
Interestingly, functional analysis indicated that Upk3b, Msln,Efemp1, and C3 positive epicardial cells were all enriched in ECM signaling. Furthermore, distinct transcriptional profiles were observed between Msln+and Efemp1+epicardial cells and Msln+and Efemp1+non-epicardial cells. The DEGs in both Msln+and Efemp1+epicardial cells were enriched in ECM associated signaling pathways, including focal adhesion, ECM organization, and extracellular structure organization. These findings highlight the important role of the ECM in the mature stage of epicardial cells.Few studies have explored the relationships between these genes and ECM signaling. Huang et al. [73] has found that givinostat, a compound used to inhibit hepatic fibrosis,effectively decreases the mRNA expression of Upk3b and Msln. After inhibition of C3 activation,mice display less glomerulosclerosis,owing to a decrease in ECM components, including collagens I, III, IV, and VI [74]. These results indicate possible relationships among Upk3b+, Msln+, C3+epicardial cells, and ECM signaling.
Adult mammals have limited ability to regenerate heart tissue after injury, because of the limited number of progenitor cells and limited proliferative capacity of cardiomyocytes. Instead of regeneration, fibrotic tissue tends to replace the injury area. The ECM provides a microenvironment for interactions among different types of cells after cardiac injury.A recent study has suggested that EPDCs differentiate into fibroblasts and smooth muscle cells,rather than cardiomyocytes or endothelial cells, after MI, and the paracrine factors secreted by EPDCs significantly improve angiogenesis and heart function, thus indicating the difference between embryonic epicardial cells and mature epicardial cells[4].Fibronectin,a major component of the ECM, is secreted by epicardial cells and plays a major role in myocardial regeneration[75].However,recent studies have suggested that the ECM may act as a barrier for pluripotency of progenitor cells[50,51].Therefore,the identification of new gene markers of mature epicardial tissue may aid in further exploration of the detailed mechanism of heart development and regeneration.
4.4. Clinical perspective
Adult epicardium has been identified as a potential therapeutic target for heart regeneration in recent years [76]. Several studies have suggested the important role of Thymosin beta 4 in cardiac repair through targeting adult epicardium [7,77,78]. A bioengineered epicardial patch with human Fstl1 protein has been found to improve the proliferation of cardiomyocytes and cardiac functions, and to decrease infarct size after MI [79]. Furthermore,human pluripotent stem cell-derived epicardial-like cells improve the contractility and calcium handling of engineered heart tissue,and enhance cardiomyocyte proliferation rate, graft size, and vascularization after cotransplantation with cardiomyocytes in infarcted hearts [80]. However, the detailed mechanisms underlying how epicardial cells contribute to cardiac repair have not been fully explored, thus hindering improvements in the clinical treatment of cardiac diseases. This study demonstrated that Upk3b,Msln, and C3 are specific gene markers of mature epicardial cells,and ECM signaling involved in these cell clusters. These findings suggest new insights regarding epicardial cells,and novel potential therapeutic targets for further research in heart regeneration,thus potentially improving clinical practice for cardiac diseases,such as MI and myocardial damage caused by various factors.
4.5. Study strength and limitations
This study identified novel potential gene markers of mature epicardium, including Upk3b, Upk1b, Efemp1, Msln, Tmem151a,Lrrn4,Rspo1,and C3,in agreement with recent studies[37,43,81].A study by Knight-Schrijver et al. [34] has also identified Upk3b and Msln as gene markers of epicardial cells. Other studies have reported high expression of Upk3b, Msln, and C3 in mature epicardium [38,82,83]. However, these studies did not specifically compare the expression of Upk3b,Msln,and C3 between embryonic and mature epicardium, nor did they identify these genes as potential markers of mature epicardium.Our results provide insights into the single-cell transcriptional and spatial characteristics of Upk3b,Upk1b,Efemp1,Tmem151a,Lrrn4,Rspo1,Msln,and C3 in both embryonic and mature epicardium. We observed significant expression of these genes in mature epicardium, thus indicating their crucial roles in the development and function of mature epicardium.Furthermore,functional analysis revealed that Upk3b+,Efemp1+, Msln+, and C3+epicardial cells were enriched in ECM signaling, thereby suggesting their potential important roles in mature epicardium through ECM signaling pathways.These results may provide novel potential targets and ideas for future basic and clinical research on epicardial cells. However, this study has some potential limitations. The 10x Genomics spatial transcriptome technology used in each Visium spot has a diameter of 55 μm,which can capture approximately 1-15 cells. However, this resolution is insufficient to distinguish between expression in epicardial and subepicardial cells. Although novel gene markers of epicardial cells and their possible functions were identified and analyzed,the exact roles of these subpopulation cells during heart development remain unclear. More studies are needed to explore the complicated functions of these cell subpopulations in heart development. Furthermore, the expression and functions of subpopulations of cells with novel gene markers(Upk3b,Efemp1,Msln,C3, etc.) were not investigated under pathological conditions.
5. Conclusions
The present study suggested clear cellular and temporal heterogeneity in epicardial cells during development. Upk3b, Efemp1,Msln,C3,etc.were identified as markers of mature epicardium,and ECM signaling was found to play a critical role in the mature epicardium.
CRediT author statement
Jianlin Du:Conceptualization, Methodology, Investigation,Writing - Reviewing and Editing, Project administration, Funding acquisition;Xin YuanandHaijun Deng:Methodology, Validation,Formal analysis;Rongzhong Huang,Liu Bin,Tianhua Xiongand
Xianglin Long:Methodology;Ling Zhang:Methodology,Software,Data curation, Writing - Reviewing and Editing;Yingrui Li:Investigation, Data curation, Writing - Original draft preparation, Visualization;Qiang She:Conceptualization, Investigation, Resources,Writing - Reviewing and Editing, Supervision, Project administration, Funding acquisition.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
Tbx18: Cre mice were donated from Professor Sylvia M. Evans.We thank Dr. Andreas Kispert from Hannover Medical School(MHH) for providing probe plasmids of Tbx18. The computational work in this paper received partial support from the Supercomputing Center of Chongqing Medical University.This work was supported by grants from the National Natural Science Foundation of China (Grant No.: 82270281), Chongqing Medical University Program for Youth Innovation in Future Medicine (Grant No.:W0133), Senior Medical Talents Program of Chongqing for Young and Middle-aged, China (Program No.: JianlinDu [2022]), Postdoctoral Research Funding of the Second Affiliated Hospital of Chongqing Medical University, China (Grant No.: rsc-postdoctor114), and Kuanren Talents Program of the Second Affiliated Hospital of Chongqing Medical University, China (Program No.:kryc-gg-2102).
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2023.07.011.
 Journal of Pharmaceutical Analysis2023年8期
Journal of Pharmaceutical Analysis2023年8期
- Journal of Pharmaceutical Analysis的其它文章
- Single-cell and spatially resolved omics: Advances and limitations
- Preface for Special Issue: Single-Cell and Spatially Resolved Omics
- Single-cell transcriptome analysis uncovers underlying mechanisms of acute liver injury induced by tripterygium glycosides tablet in mice
- A single-cell landscape of triptolide-associated testicular toxicity in mice
- Temporal dynamics of microglia-astrocyte interaction in neuroprotective glial scar formation after intracerebral hemorrhage
- Promise of spatially resolved omics for tumor research