
健脾扶正颗粒HPLC 指纹图谱的建立及8 个化学成分含量测定
2023-09-25 07:24:28李晓玲石雨荷侯超文刘湘丹童巧珍
湖南中医药大学学报 2023年9期
聂 格,李晓玲,石雨荷,李 晴,朱 珏,侯超文,刘湘丹,童巧珍*
1.湖南中医药大学,湖南 长沙 410208;2.浏阳市中医医院,湖南 浏阳 410300;3.湖南中医药大学附属第二中西医结合医院,湖南 浏阳 410300
功能性消化不良是临床常见的一种功能性胃肠疾病,是指经标准检查后未发现器质性疾病而无法解释的消化不良的统称[1-2],该病严重威胁患者的生活质量[3]。 健脾扶正颗粒为浏阳市中医医院的制剂,该方由太子参、黄芪、茯苓等9 味中药材组成,方中诸药紧扣脾胃虚弱,气机阻滞,升降失司这一基本病机[4-5],临床常用于功能性消化不良证。 其中太子参益气健脾、生津润肺[6-7]。 黄芪补气升阳、固表止汗、生津养血[8],主要成分为黄芪甲苷、毛蕊异黄酮糖苷、芒柄花素[9],经现代药理学证明其有免疫调节、抗肿瘤,促进胃排空的作用[10-11]。 茯苓利水渗湿、健脾宁心,所含的成分茯苓酸被认为能抗肿瘤、镇静催眠[12-13]。 桔梗宣肺化痰、利咽排脓[14],桔梗皂苷D是其中含量相对较高的一种成分,在抗肿瘤、调节免疫等方面均表现出良好的药理活性[15]。 五味子益气生津、补肾宁心[16],其中五味子醇甲可改善腹泻[17],加强免疫等多重功效。 该方在浏阳市中医医院经过数千例临床应用,疗效确切,制成的颗粒剂更加方便患者的携带。
本实验通过检测15 批健脾扶正颗粒样品,建立其HPLC 指纹图谱,并对复方中的8 种指标性成分进行了含量测定,为该制剂的临床用药提供了保障。
1 材料
1.1 仪器
1260 Infinity LC 高效液相色谱仪(美国安捷伦公司);Waters Arc-2-型蒸发光散射检测器(美国沃特世公司);ME204E 型电子天平[梅特勒-托利多仪器(上海)公司];Waters Sun fire C18色谱柱(250 mm×4.6 mm,5 μm);DZKW-S-6 型电热恒温水浴锅(北京市永光明医疗仪器有限公司);98-1-B 型电子调温电热套(天津市泰斯特仪器有限公司);SB25-12DT 型数控超声波清洗器(宁波新芝生物科技股份有限公司);RE-2000A 型旋转蒸发器(上海亚荣生化仪器厂)。
1.2 试药
健脾扶正颗粒(浏阳市中医医院的临床经验方),批号200505、200614、200714、200903、201110、210108、210208、210314、210419、210610、210725、211116、211117、220108、220109 (S1 ~S15), 黄 芪甲苷对照品(纯度≥98%,批号PS012327)、毛蕊异黄酮糖苷对照品(纯度≥98%,批号PS000687)、芒柄花素对照品(纯度≥98%,批号PS000674)、桔梗皂苷对照品(纯度≥98%,批号PS010786)、五味子醇甲对照品(纯度≥98%,批号PS011670)、茯苓酸对照品(纯度≥98%,批号PS011828)、太子参环肽B对照品(纯度≥98%,批号PS000879)、葫芦巴碱对照品(纯度≥98%,批号PS012765)均购自成都普思生物科技股份有限公司,乙腈、磷酸为色谱纯,水为怡宝水,其他试剂为分析纯。
2 方法与结果
2.1 色谱条件
2.1.1 HPLC-UV 色谱条件 以Waters Sun fire C18(250 mm×4.6 mm,5 μm)为色谱柱;以乙腈为流动相A,0.1%磷酸水为流动相B,梯度洗脱:0~8 min,2%→15% A;8~26 min,15%→44% A;26~28 min,44% A;28~30 min,44%→45% A;30~33 min,45%→50% A;33~42 min,50%→64% A;42~51 min,64%→79% A;51~63 min,79%→98% A;63~68 min,98% A;流速为1.0 mL·min-1;柱温为30 ℃;检测波长为203 nm、210 nm、250 nm 多波长切换;进样体积为10 μL。
2.1.2 HPLC-ELSD 蒸发光散射条件 以Waters Sun fire C18(250 mm×4.6 mm,5 μm)为色谱柱;以水为流动相A,乙腈为流动相B,梯度洗脱:0~5 min,2%→15.7% B;5~10 min,15.7%→29.4% B;10~15 min,29.4%→43.1% B;15~17 min,43.1% B;17~18 min,43.1%→45.9% B;18~19 min,45.9%→54.1% B;19~35 min,54.1%→98%B;35~36 min,98%→2% B;体积流量1.0 mL·min-1;柱温30 ℃;ELSD 增益60,漂移管温度65 ℃,气体压力为35psi,进样量10 μL。
2.2 溶液的制备
2.2.1 对照品溶液的制备 取黄芪甲苷、毛蕊异黄酮糖苷、芒柄花素、桔梗皂苷D、五味子醇甲、茯苓酸、太子参环肽B、葫芦巴碱对照品适量,精密称定,加入甲醇制成每毫升含0.38 mg、0.46 mg、0.84 mg、0.48 mg、0.97 mg、0.88 mg、0.58 mg、0.90 mg 的混合标准溶液,0.22 μm 微孔滤膜滤过,即得。
2.2.2 供试品溶液的制备 取健脾扶正颗粒约取6 g,精密称定,研磨至细粉,过筛(6 号筛),置500 mL 圆底烧瓶中,加入80%甲醇150 mL 加热回流3 h,蒸干提取液,残渣用150 mL 水溶解,用水饱和的正丁醇萃取3 次,每次50 mL,合并萃取液,蒸干,加80%甲醇25 mL 溶解,摇匀称定,用80%甲醇补足,经微孔滤膜(0.45 μm)滤过,取续滤液,即得。 单味药材溶液的制备分别取处方量的各药材,精密称定,打成粗粉,过筛(2 号筛),分别加入甲醇50 mL 放入100 mL 锥形瓶中,超声提取40 min 后用甲醇补足,经微孔滤膜(0.45 μm)滤过,即得。
3 方法学考察
3.1 线性关系考察
分别取毛蕊异黄酮糖苷、芒柄花素、五味子醇甲、茯苓酸、太子参环肽B、葫芦巴碱对照品溶液0.5、1.0、1.5、2.0、2.5 和3.0 mL,置10 mL 容量瓶中,加甲醇稀释至刻度,配成系列质量浓度混合对照品溶液,以进样浓度量(X)为横坐标,对应的峰面积积分值(Y)为纵坐标;分别取桔梗皂苷D、黄芪甲苷对照品溶液0.5、0.8、1.0、1.3、1.6 mL 置10 mL 容量瓶中,加甲醇稀释至刻度,以进样量微克数的自然对数为横坐标,以桔梗皂苷D、黄芪甲苷色谱峰面积的自然对数为纵坐标,进行线性回归得标准曲线方程与线性范围见表1。

表1 线性关系
3.2 精密度试验
分别精密吸取上述“2.2.1”项下对照品溶液10 μL,在色谱条件“2.1”下,连续进样6 次,黄芪甲苷、毛蕊异黄酮糖苷、芒柄花素、桔梗皂苷D、五味子醇甲、茯苓酸、太子参环肽B、葫芦巴碱各标准品出峰的保留时间RSD(n=6)≤0.90%,各峰面积的RSD(n=6)分别为2.17%、0.85%、0.77%、2.48%、0.92%、0.81%、0.64%、0.94%,结果表明仪器精密度良好。
3.3 重复性试验
分别取同一批健脾扶正颗粒(批号200614) 6份,精密称定,按“2.2.2”项下操作,在色谱条件“2.1”下测定,分别计算8 个对照品黄芪甲苷、毛蕊异黄酮糖苷、芒柄花素、桔梗皂苷D、五味子醇甲、茯苓酸、太子参环肽B、葫芦巴碱各标准品出峰的保留时间RSD(n=6)≤0.33%,峰面积的RSD(n=6)分别为1.79%、2.42%、2.17%、2.98%、2.68%、2.40%、2.40%、2.71%,表明方法的重复性良好。
3.4 稳定性试验
分别取同一批健脾扶正颗粒(批号200614)6份,精密称定,按“2.2.2”项下操作,色谱条件“2.1”下测定,分别于室温下放置0、2、4、8、16、24 h,其中8个对照品黄芪甲苷、毛蕊异黄酮糖苷、芒柄花素、桔梗皂苷D、五味子醇甲、茯苓酸、太子参环肽B、葫芦巴碱的各标准品出峰的保留时间RSD(n=6)≤0.42%,峰面积RSD(n=6)分别为2.84%、1.03%、0.53%、2.02%、1.07%、0.74%、0.70%、1.10%,说明供试品在24 h 内稳定性良好。
3.5 加样回收率试验
分别取已知含量的健脾扶正颗粒样品(批号200614)6 份,精密称定,分别精密加入与样品中成分含量接近的对照品黄芪甲苷、毛蕊异黄酮糖苷、芒柄花素、桔梗皂苷D、五味子醇甲、茯苓酸、太子参环肽B、葫芦巴碱对照品溶液,按照“2.2.2”项下操作,在色谱条件“2.1”下测定并计算加样回收率,计算得到平均回收率(n=6)分别为100.93%、95.93%、100.79%、104.59%、101.49%、96.26%、99.98%、100.45%。 RSD(n=6)分别为0.97%、1.44%、1.48%、1.01%、1.20%、1.20%、2.38%、2.15%。
4 指纹图谱
4.1 指纹图谱的建立
分别取15 批健脾扶正颗粒(S1~S15),按“2.2.2”项下操作,色谱条件“2.1”下测定,将测得的15 批HPLC 色谱图导入《中药色谱指纹图谱相似度评价系统》(2012A 版),得到样品指纹图谱叠加图,结果见图1A、图2A。 对15 批健脾扶正颗粒的指纹图谱进行相似度计算,HPLC-UV 结果范围为0.996~0.999。 HPLC-ELSD 结果范围为0.948~0.999。 相似度均在0.90 以上,说明健脾扶正颗粒质量稳定,化学成分一致性较好,但各成分含量存在差异。 以S1作为参照图谱,对15 批样品的相关参数进行多点校正和Mark 峰匹配,生成对照图谱,选择吸收信号较强、峰形明显、稳定性较好的共有峰进行标定,标得33 个共有峰,结果见图1B、图2B。

图1 15 批健脾扶正颗粒指纹图谱叠加图(A)、样品(B)、对照品(C)HPLC-UV 图
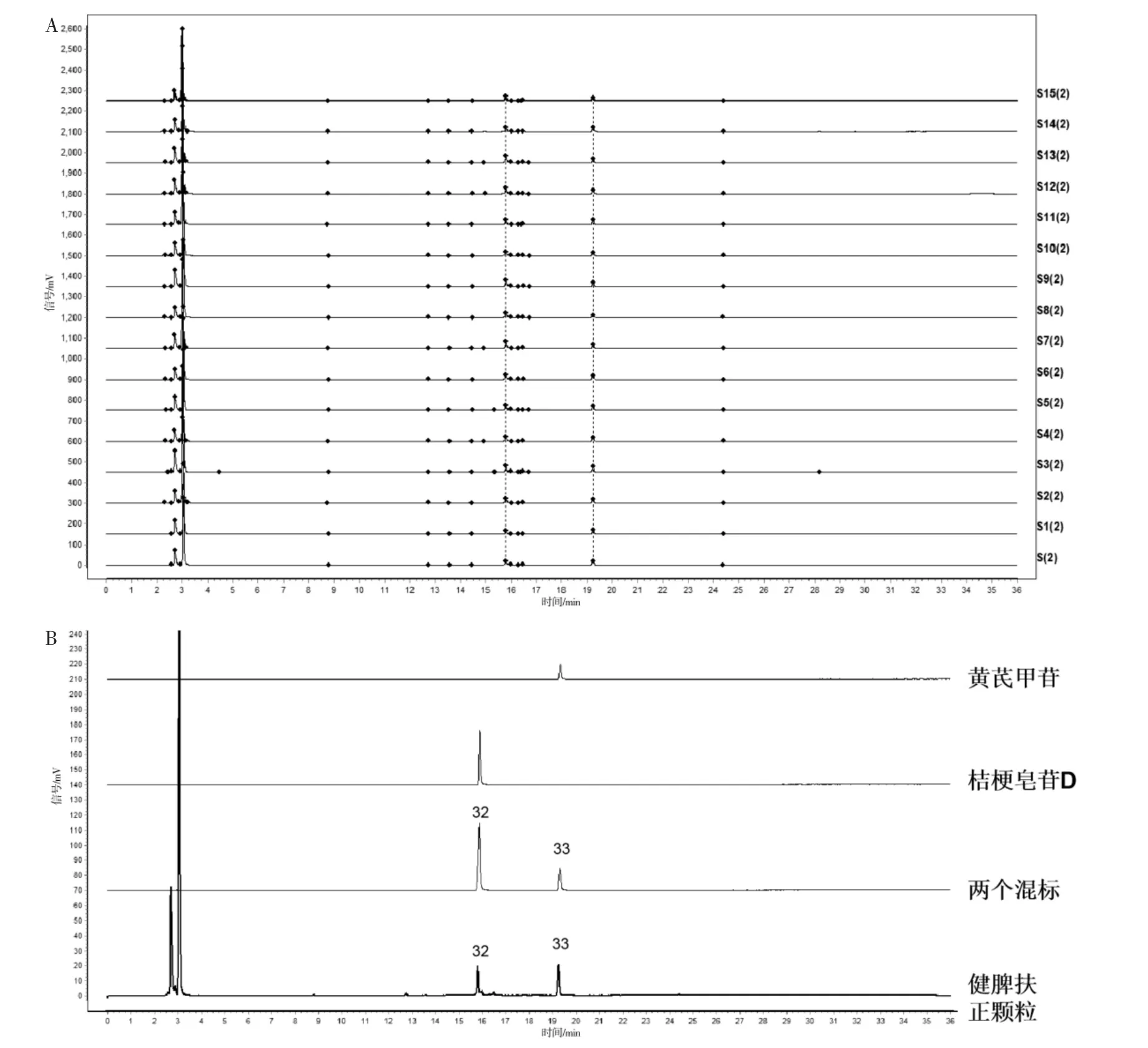
图2 15 批健脾扶正颗粒指纹图谱叠加图(A)、样品和对照品峰(B)HPLC-ELSD 图
4.2 共有峰的指认及归属
按照“2.1.1”项下色谱条件分别测定供试品溶液、对照品溶液、药材溶液,通过对比各峰保留时间,对各峰进行指认,确认了6 个色谱峰,分别为1 号峰(葫芦巴碱)、6 号峰(毛蕊异黄酮糖苷)、14 号峰(太子参环肽B)、16 号峰(芒柄花素)、18 号峰(五味子醇甲)、30 号峰(茯苓酸)。按照“2.1.2”项下色谱条件分别测定供试品溶液和对照品溶液,确认了2 个色谱峰,依次是32 号峰(桔梗皂苷)、33 号峰(黄芪甲苷)。1 号峰来自白扁豆,3、18、19、20、21、22、24、25、26、27、28、29 号峰来自五味子,4、5、14、31 号峰来自太子参,2、6、8、10、12、16 号峰来自黄芪,7、9、11、30号峰来自茯苓。 其中13、15、17、23 峰的药材来源有待进一步研究。 各种色谱图详见图2—4。
4.3 含量测定
分别取15 批健脾扶正颗粒样品6 g,精密称定,按“2.2.2”项下操作。分别精密吸取对照品溶液与供试品溶液进行HPLC 测定,计算8 个成分的含量测定结果见表2。
5 化学模式识别
5.1 聚类分析(PCA)
本研究分别以健脾扶正颗粒指纹图谱中标定的33 个(包括蒸发光散射检测的2 个峰)进行聚类分析,以共有峰的相对峰面积为变量,运用SPSS 26.0统计软件,采用组间均连法,结果见图3,d=25 时,15 批样品可以分为2 类,即S1、S2、S3、S4、S5、S7、S9、S11、S13、S14 被聚为第1 类,其他被聚为第2类。d=10 时,后一类又可以聚为四类,即S6、S15 聚为一类,S8、S10、S12 聚为一类,S9、S14、S1、S7 聚为一类,S5、S13、S11、S2、S4、S3 聚为一类。

图3 供试品溶液及单个对照品溶液HPLC-UV 色谱图
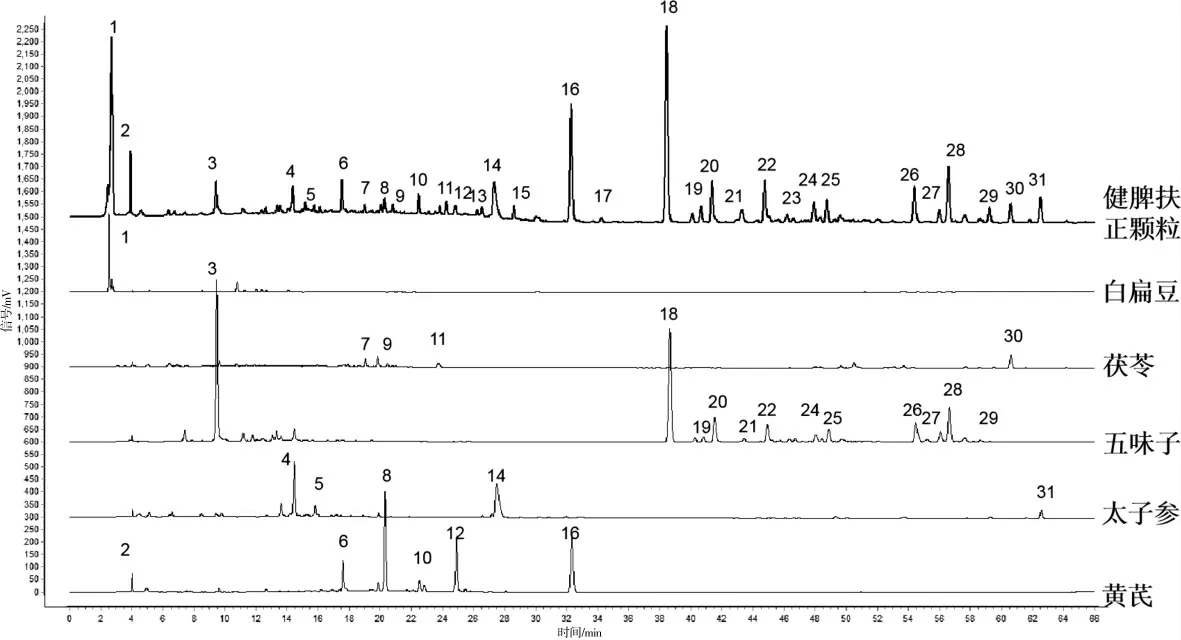
图4 供试品溶液及各单味药材提取液HPLC 图
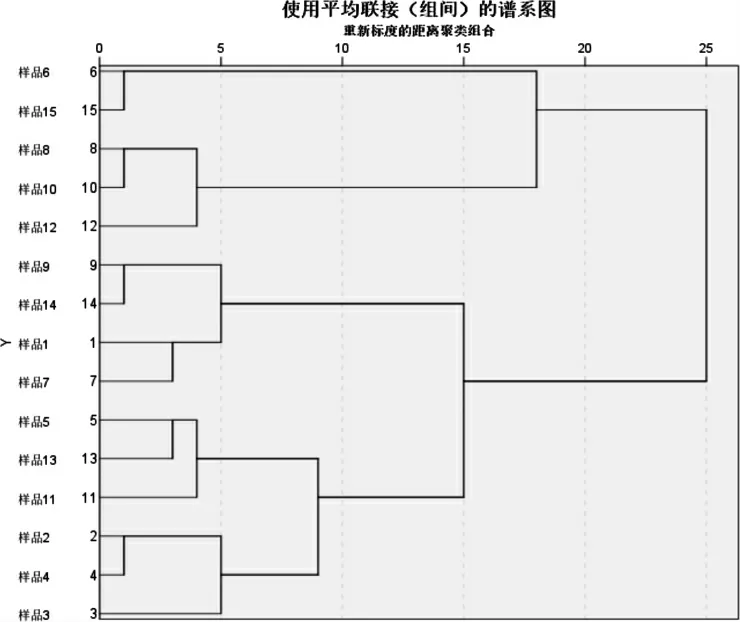
图5 健脾扶正颗粒聚类分析图
5.2 主成分分析
以各共有峰的相对峰面积为指标,将15 批健脾扶正33 个共有峰的相对峰面积进行标准化处理后,采用SPSS 26.0 软件进行主成分分析,得到各成分碎石图、主成分矩阵表计算其特征值和方差贡献率,以特征值>1.5 为标准,得到前4 个主成分因子的特征值,其累积方差贡献率为82.249%,结果见表3,图6 表明前4 个成分较陡,剩余的其他成分之间比较平缓。 选择提取该4 个主成分因子对15 批健脾扶正颗粒样品进行评分,以方差贡献率为分配系数,计算各批样品的主成分因子得分及综合得分(综合得分=相应因子得分×,综合得分越高表示样品整体质量越好,最后对综合得分进行排序,由表4 可知,不同批次健脾扶正颗粒质量高 低 为S1、S6、S4、S10、S8、S5、S15、S9、S7、S12、S2、S14、S13、S11、S3。
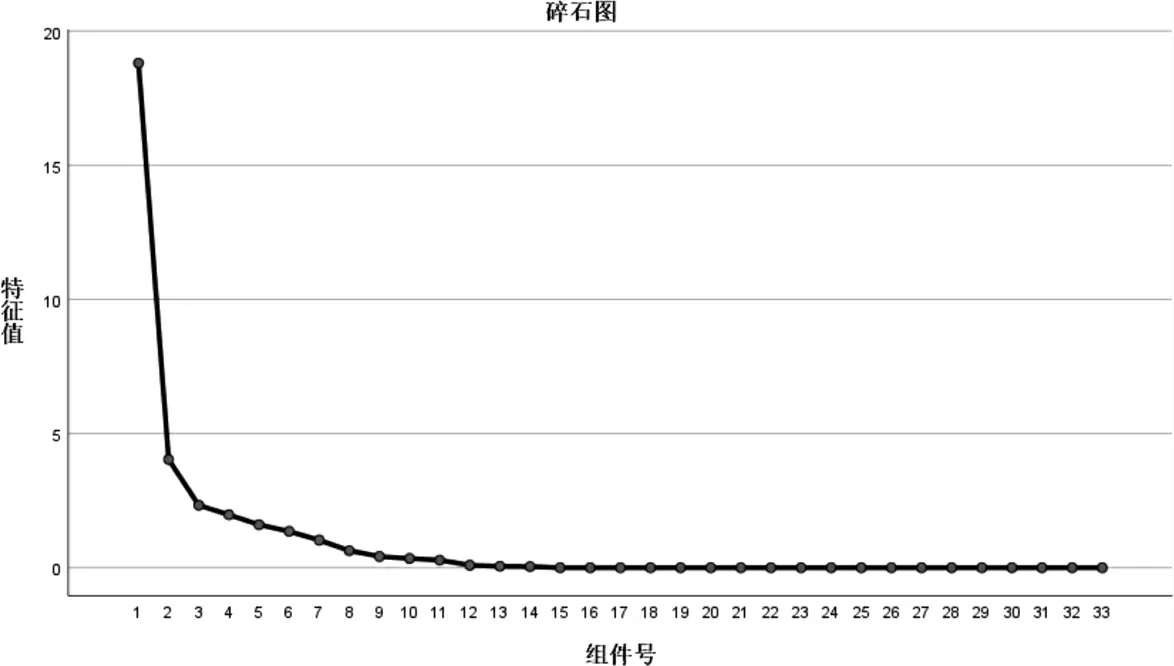
图6 主成分分析碎石图

表3 4 个主成分因子的特征值和方差贡献率
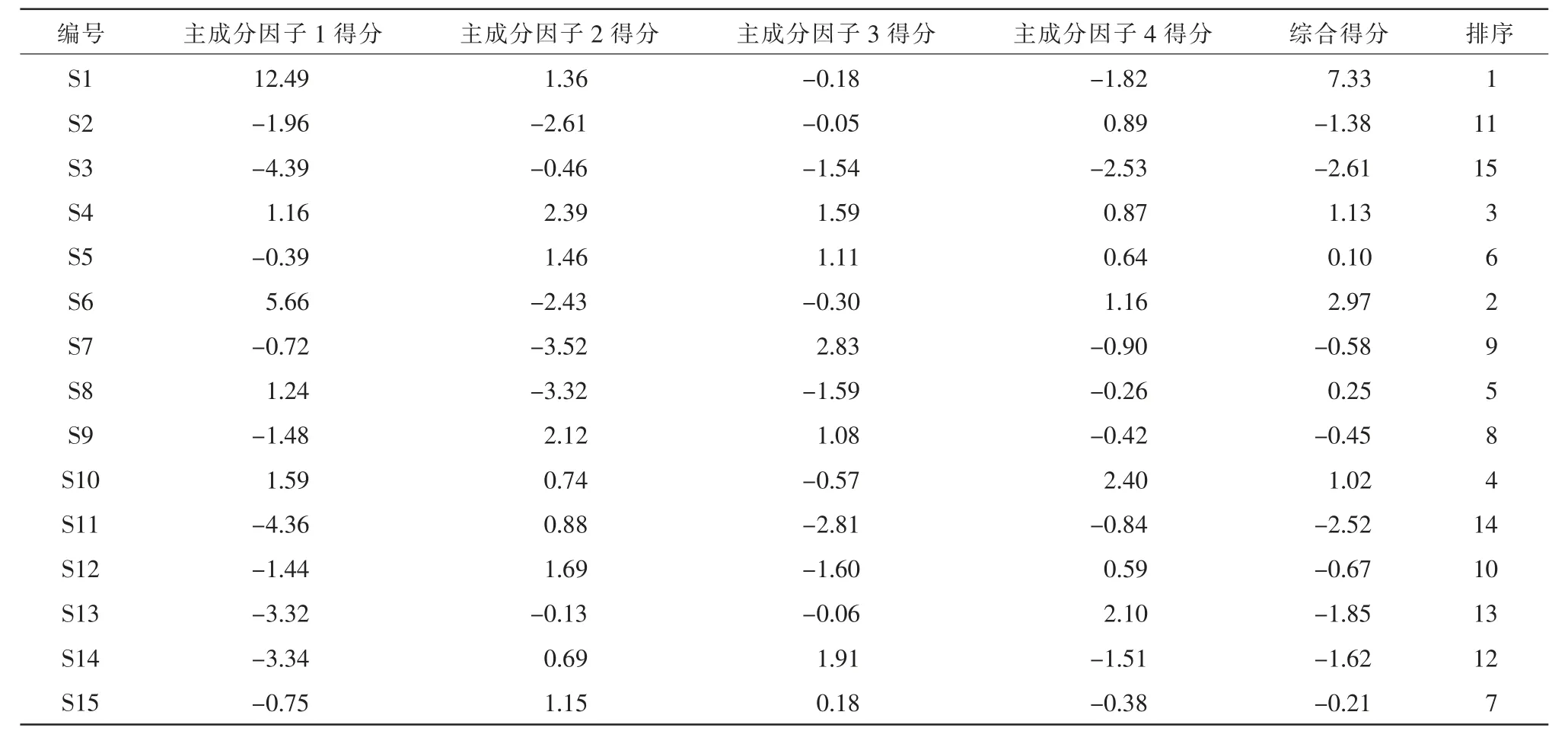
表4 15 批样品主成分因子得分及排序
6 讨论
本研究前期分别考察了不同波长(203 nm、210 nm、230 nm、250 nm、280 nm)、不同提取方法(加热回流提取,超声提取)、不同提取溶剂(甲醇、80%甲醇、50%甲醇、水)、不同流动相体系(乙腈-水,乙腈-0.1%磷酸水溶液)、不同柱温(30 ℃、40 ℃)的影响,结果发现在波长(203 nm、210 nm、250 nm)切换、80%甲醇溶剂加热回流提取、0.1%磷酸水溶液-乙腈为流动相、柱温30 ℃的条件下提取的色谱峰信息较为丰富,有较好的色谱峰峰形,其中黄芪甲苷和桔梗皂苷不适用于在紫外下测定,故采用蒸发光散射检测器进行检测。
本研究建立的指纹图谱,对照指纹图谱的相似度均大于0.90,共标定了33 个共有峰,其中指认了8 个色谱峰。通过对15 批健脾扶正颗粒进行含量测定,显示这15 批样品8 个标准品黄芪甲苷、毛蕊异黄酮糖苷、芒柄花素、桔梗皂苷D、五味子醇甲、茯苓酸、太子参环肽B、葫芦巴碱含量分别为0.26~0.30、0.07~0.08、0.27~0.35、0.15~0.17、0.40~0.50、0.27~0.34、0.17~0.22、0.23~0.32 mg·g-1,表明健脾扶正颗粒制备工艺稳定,质量可控性良好。 近年来,研究人员通过指纹图谱结合聚类分析和主成分分析,对中药材制剂的质量进行更加全面的评价[18-19]。 本实验以共有峰的相对峰面积为变量得到的结果显示S1、S2、S3、S4、S5、S7、S9、S11、S13、S14 被聚为1 类,其他被聚为第2 类,以S1 样品的综合得分最高。 综上所述,本研究建立的指纹图谱及聚类分析和主成分分析结果可为健脾扶正颗粒剂的质量控制提供参考。
猜你喜欢
西北药学杂志(2023年4期)2023-07-27 07:19:14
Journal of Traditional Chinese Medicine(2022年3期)2022-07-20 15:54:08
Journal of Traditional Chinese Medicine(2022年3期)2022-07-20 15:53:38
中医药导报(2021年4期)2021-11-22 12:21:12
花卉(2021年14期)2021-07-26 02:22:38
世界科学技术-中医药现代化(2018年9期)2019-01-29 03:40:50
养生保健指南(2018年3期)2018-04-13 09:21:02
邵阳学院学报(自然科学版)(2015年2期)2015-06-05 12:22:39
中国刑警学院学报(2014年4期)2014-04-27 10:08:00
中国中医药现代远程教育(2014年22期)2014-03-01 04:32:48
