
液相预氧化聚丙烯腈纤维的制备及性能研究
2023-09-20 03:17胡世棋葛建龙刘其霞王玉萍
棉纺织技术 2023年9期
胡世棋 葛建龙 季 涛 刘其霞 王玉萍
(1.南通大学,江苏南通,226019;2.江苏苏通碳纤维有限公司,江苏南通,226005)
聚丙烯腈(PAN)基活性炭纤维的制备过程包括预氧化、炭化和活化,其中预氧化过程是制备PAN 基炭纤维和活性炭纤维的重要步骤[1]。根据介质不同可以分为液相预氧化、气相预氧化以及固相预氧化。固相预氧化应用较少,相关研究报道极少;气相预氧化操作简单,是目前较为成熟的预氧化工艺,但其处理的时间较长,且加热温度较高,是制备PAN 基活性炭纤维过程中耗时最久的工艺[2-4];液相预氧化反应较温和,预氧化处理时间短、温度低,所制备的预氧化纤维结构均匀,不产生皮芯结构[5-6]。
本研究通过液相预氧化法制备出预氧化PAN 纤维,通过傅里叶红外光谱(FT-IR)、热重分析(TG)、差示扫描量热法(DSC)、拉伸强度等指标对预氧化PAN 纤维进行表征分析,以期为高力学性能预氧化纤维的制备提供参考。
1 试验部分
1.1 试验原料及仪器
原料:PAN(日本东丽株式会社),乙二醇,碳酸胍,N-羟基邻苯二甲酰胺,无水乙醇(上海麦克林生化科技有限公司),超纯水(自制)。
仪器:HH-2DS 型油浴锅,DHG 型电热鼓风干燥箱,Nicolet IS 50 型傅里叶红外光谱仪,Gemini SEM 300 型场发射扫描电镜,DSC200 F3 Maia 型差示扫描量热仪,TG209 F3 型热重分析仪,XJ810 型电子万能材料试验机。
1.2 液相预氧化PAN 纤维的制备
将乙二醇、碳酸胍和N-羟基邻苯二甲酰亚胺按照一定比例混合,称取一定质量的PAN 原丝,通过改变预氧化时间和预氧化温度进行液相预氧化,原丝记为P0。固定预氧化时间为90 min,分别在预氧化温度为180 ℃、190 ℃、200 ℃、210 ℃、220 ℃、230 ℃对PAN 进行液相预氧化,将相应制备的预氧化PAN 纤维样品分别记为P180、P190、P200、P210、P220、P230。固 定 预 氧 化 温 度 为220 ℃,分别在预氧化时间10 min、20 min、30 min、40 min、50 min、60 min、70 min、80 min、90 min 对PAN 进行液相预氧化,将制备的预氧化PAN 纤维样品分别记为P10、P20、P30、P40、P50、P60、P70、P80、P90。
1.3 测试与表征
采用Gemini SEM 300 型场发射扫描电镜观察不同预氧化条件下的纤维纵向形态。
采用DSC200 F3 Maia 型差示扫描量热仪对不同预氧化条件下的纤维热转变性能进行分析,升温速率为5 ℃/min。
采用TG209 F3 型热重分析仪对不同预氧化条件下的纤维热稳定性进行分析,升温速率为20 ℃/min。
采用Nicolet IS 50 型红外光谱仪对不同预氧化条件下的纤维化学结构进行表征,并计算不同预氧化条件下的纤维环化率[7]。
采用XJ810 型电子万能材料试验机对不同预氧化条件下的纤维力学性能进行测试,夹持距离为2 cm,下降速度为5 mm/min。
2 结果与讨论
2.1 液相预氧化纤维形貌
由图1PAN 原丝和液相预氧化PAN 纤维的扫描电镜照片可知,PAN 原丝表面光滑有残留杂质,经过液相预氧化处理后,纤维的表面仍保持光滑完整的状态,没有出现沟槽,并去除了原丝表面残留的油剂和其他杂质,说明液相预氧化反应较温和,对纤维的损伤较小。此外,通过观察PAN原丝与各处理条件下的预氧化PAN 纤维宏观形态,发现随着预氧化温度的升高和预氧化时间的延长,纤维颜色都发生由白色到红棕色到深褐色再到黑色的转变,当预氧化温度为220 ℃,预氧化时间为90 min 时纤维已经完全变黑,这种颜色变化可以认为源于聚合物中形成了甲亚胺、多烯之类的发色基团结构[8]。纤维的颜色全变为黑色被认为是已经形成了热稳定结构的标志,说明此时的环化程度较高[9]。在预氧化过程中,PAN 纤维会发生环化、氧化及脱氢等一系列反应,分子结构中的C≡N 基团以环化的形式发生各种结构转变,形成高稳定性的梯形结构,在脱氢反应和氧化反应中还会发生其他结构转变,从而形成含有C=C 和C=N 的 共轭结构[10]。

图1 PAN 原丝和液相预氧化PAN 纤维的SEM 图
2.2 液相预氧化PAN 纤维红外光谱分析
图2 为PAN 原丝与预氧化温度为220 ℃,预氧化时间为90 min 的液相预氧化纤维的FT-IR谱图。
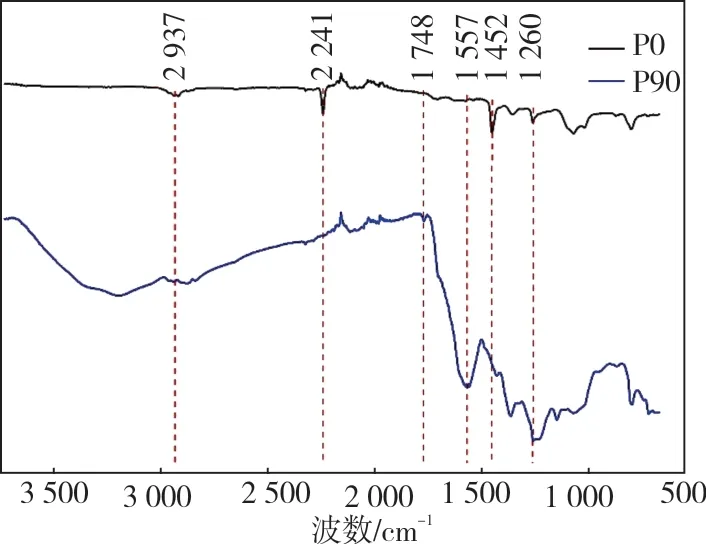
图2 聚丙烯腈原丝与液相预氧化纤维FT-IR 图
由图2 可以看出,经过液相预氧化处理的纤维结构发生了较大的变化,1 557 cm-1处对应于C=N 伸缩振动吸收峰的强度显著增强,同时2 241 cm-1处对应的C≡N 伸缩振动吸收峰变得很不明显,表明预氧化过程中发生了环化反应,PAN 线型分子转变为环状梯形结构,并且结构转变比较完全。2 937 cm-1处对应于C—H 伸缩振动吸收峰和1 452 cm-1对应于C—H 面内弯曲振动吸收峰的强度减小,1 260 cm-1处对应于C—H面内弯曲振动吸收峰强度增加,表明预氧化过程中发生了脱氢反应。1 748 cm-1处出现的对应于C=O 伸缩振动吸收峰,是由于预氧化过程中发生氧化反应,在预氧化反应后期氧化反应形成了不饱和结构。可见,预氧化过程中至少发生了环化、脱氢和氧化三种化学反应[11-12]。
图3 和图4 分别为不同预氧化温度和不同预氧化时间下预氧化PAN 纤维的环化率。

图3 预氧化温度对纤维环化率的影响

图4 预氧化时间对纤维环化率的影响
从图3 中可以看出,在氧化时间为90 min 保持不变时,预氧化PAN 纤维环化率随着氧化温度的升高,呈现先升高后下降的趋势,在220 ℃时达到最高为46.8%,说明220 ℃以后环化反应基本结束,此时预氧化PAN 纤维的环化程度最高,当温度继续升高,部分分子链由于耐热性能较差,会发生裂解产生HCN 以及CO2,使得环化率下降[13]。从图4 可以看出,在氧化温度为220 ℃保持不变时,预氧化PAN 纤维的环化率随着预氧化时间的延长而升高,这是由于随着时间的延长,PAN 线型分子链上氰基的环化反应发生越多,使得预氧化丝的梯形结构也就越多,即环化程度越大,这与相关研究结果一致[14],在90 min 时环化率达到最大46.8%。结合图3 和图4 可知,氧化温度220 ℃,氧化时间90 min 时,预氧化PAN 纤维的环化率达到最高46.8%。
2.3 液相预氧化PAN 纤维热重分析
图5 是PAN 原丝和不同氧化温度下制备的预氧化PAN 纤维的TG 曲线(预氧化时间90 min)。图6 为PAN 原丝和不同预氧化时间下制备的预氧化PAN 纤维的TG 曲线(预氧化温度220 ℃)。

图5 原丝和不同预氧化温度下制得的预氧化PAN 纤维TG 曲线

图6 原丝和不同预氧化时间下制得的预氧化PAN 纤维TG 曲线
从图5 中可以看出,PAN 原丝及液相预氧化PAN 纤维在300 ℃之前分解速度相对较慢,质量分数下降幅度较小(约10%以内),基本保持稳定;而在300 ℃到500 ℃之间,质量分数大幅下降,主要是预氧化阶段未完全环化交联的氰基在此阶段继续反应,另外氰基环化放出的热量又会使已形成梯形结构的末端氨基以NH3的形式脱出,从而引起明显的热失重[15]。在预氧化时间90 min条件下,随着预氧化温度的升高,预氧化PAN 纤维残炭率逐渐增加,在220 ℃达到最高,此时残炭率为57.7%,环化程度也最高,这与环化率结果一致。从图6 中可以看出,在预氧化温度为220 ℃的条件下,随着预氧化时间的延长,纤维质量分数不断下降,液相预氧化PAN 纤维的残炭率均高于原丝,到800 ℃时所有预氧化PAN 纤维的残炭率在52%~58%范围内[16]。
2.4 液相预氧化纤维差示扫描量热分析
图7 是PAN 原丝和不同预氧化温度下制得的预氧化PAN 纤维DSC 曲线(预氧化时间90 min)。图8 是PAN 原丝和不同预氧化时间下制得的预氧化PAN 纤维DSC 曲线(预氧化温度220 ℃)。
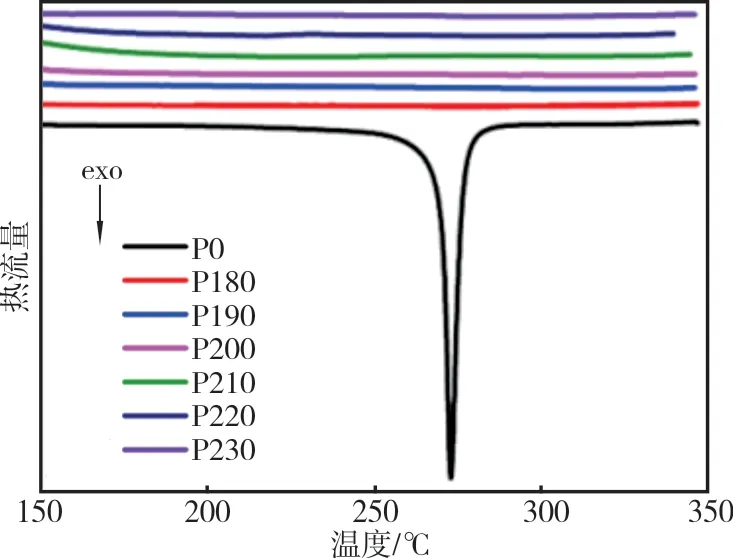
图7 原丝和不同预氧化温度下制得的预氧化PAN纤维DSC 曲线

图8 原丝和不同预氧化时间下制得的预氧化PAN纤维DSC 曲线
从图7 可以看出,原丝在272 ℃出现明显的放热峰,这是由于氰基发生环化放热反应引起的,放热反应起始于242 ℃,随后放热反应迅速发展,并在272 ℃时放热量达到最大,在286 ℃时放热过程基本结束。不同温度下制得的预氧化PAN 纤维DSC 曲线平稳,没有出现放热峰,说明在预氧化时间90 min 时,不同温度下制得的预氧化PAN 纤维已发生环化反应。
从图8 可以看出,与原丝相比氧化时间为10 min 制得的预氧化PAN 纤维的放热峰在较低温度下出现且变宽,放热反应起始于225 ℃,随后在260 ℃时放热量达到最大,在288 ℃时放热结束,说明预氧化致使环化起始温度提前,加速了环化反应的发生。
综上所述,液相预氧化降低了反应初始温度,且在温和的条件下获得较好的环化程度,提高了预氧化纤维的热稳定性[17-18]。
2.5 液相预氧化纤维力学性能分析
图9 为预氧化时间和预氧化温度对预氧化PAN 纤维强力的影响。

图9 预氧化温度和预氧化时间对预氧化PAN 纤维强力的影响
从图9(a)中可以看出,在预氧化时间保持90 min 不变的条件下,随着预氧化温度的升高,预氧化PAN 纤维的强力逐渐下降,这是由于预氧化温度升高,纤维的结构被破坏,且温度越高,反应程度越剧烈。200 ℃到210 ℃条件下,预氧化PAN 纤维的强力下降最明显,210 ℃以后,纤维的强力呈缓慢下降的趋势,这是由于氧化过程使得纤维中大分子链沿纤维轴向的取向度下降,分子链的排列逐渐趋于无序化,导致其强力下降[19]。
图9(b)为预氧化温度220 ℃,不同预氧化时间对纤维强力的影响。可以看出,强力保留率随着氧化时间的延长呈现出先下降再上升,后逐渐趋于平缓的趋势。经过10 min~30 min 氧化后纤维强力保留率呈现下降,主要是因为预氧化时间较短,原丝中柔性大分子链变得僵硬,环化程度较低[20]。预氧化时间超过30 min 后,纤维的强力保留率逐渐升高并趋于稳定,这是由于随着氧化时间延长,预氧化过程中参与环化反应的分子数增多,分子交联度不断增加,使得纤维逐渐形成环化结构,因而强力保留率不断升高;当预氧化时间超过70 min 后,环化反应基本结束,因而强力保留率趋于稳定[21];预氧化温度为220 ℃,预氧化时间为90 min 时,纤维的强力保留率为76.1%。
3 结论
(1)采用液相预氧化法所制备的液相预氧化纤维表面光滑,去除了油剂以及多余的杂质,未出现皮芯层结构和破损现象。
(2)液相预氧化法降低了预氧化纤维环化反应初始温度,加速了环化反应的发生,且在温和的条件下获得了的较好环化程度。
(3)在预氧化温度为220 ℃,预氧化时间为90 min 的条件下所获得的预氧化PAN 纤维的热稳定性最好,此时预氧化纤维的环化率为46.8%,残炭率为57.7%,强力保留率为76.1%。
猜你喜欢
纺织服装周刊(2022年32期)2022-09-08
四川劳动保障(2021年9期)2022-01-18
环境保护与循环经济(2021年7期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
今日农业(2019年15期)2019-09-03
制造技术与机床(2018年11期)2018-11-23
小哥白尼(军事科学)(2018年2期)2018-05-25
高科技纤维与应用(2016年1期)2017-01-17
河北工业科技(2015年4期)2015-02-27
食品工业科技(2014年9期)2014-03-11
