
基于宏基因组学测序技术分析3 个轮次高温大曲微生物群落
2023-09-13 02:52王国峥卢建军陈良强张巧玲
食品科学 2023年16期
王国峥,陈 笔,卢建军,陈良强,张巧玲,罗 寒
(贵州茅台股份有限公司,贵州省酿酒工程技术研究中心,贵州 遵义 564501)
曲是我国传统白酒酿造的重要原料,曲研究对于酿酒乃至整个传统酿造行业都有十分重要的意义[1-2]。酱香型白酒风味复杂,这与其发酵过程中曲用量高密切相关[2],其生产可分为下沙、造沙、1~7轮次,每个轮次都需要添加大量曲粉作为糖化剂与发酵剂,但各轮次生产因所处的环境气候不同可能导致大曲微生物群落结构与功能出现差异[3]。下沙、造沙轮次是酱香型白酒生产的投粮阶段,高粱糊化程度较低[4],1轮次在9 个轮次中环境均温最低、环境湿度最低[5],这些不利因素都会阻碍发酵进程,因此大曲的功能在前期轮次的生产中更为重要,不仅直接决定发酵的进程,也关系到后期轮次微生物代谢产物、前体物质及香味物质的积累与形成[6]。因此本研究选取这3 个生产轮次的生产用曲作为实验对象,进行微生物群落结构及功能分析。
随着现代分子生物学技术的发展,高通量测序技术逐渐被用于食品微生物的研究中[7-8],旨在全面揭示微生物群落结构与功能信息。目前对酿造微生物菌群结构及功能研究大多采用扩增子测序技术[9-15],例如Wang Li等[15]利用16S rRNA扩增子测解析茅台酒发酵过程中细菌群落动态变化。然而扩增子测序得到的分子信息较为简单[16],难以精细解析大曲微生物群落特征,而宏基因组学测序技术则是通过获取样品中全部基因序列解析微生物群落信息方法,可以准确获取大量生态信息数据。宏基因组测序已成为研究微生物多样性和群落特征的重要手段[17],相比于高通量测序技术,宏基因组测序技术可以更为全面挖掘出微生物基因组信息,便于研究者挖掘菌群的生物多样性、群落结构、功能特性和相互关系[18]。现已有一些学者将宏基因组测序应用于酿造微生物群落的研究当中[18-23],其中也可见一些高温大曲的研究,例如Fan等[18]利用宏基因组测序技术对汾酒大曲进行全面解析,发现乳酸菌是群落中的主要物种,而酿酒酵母仅为群落总数的0.005%,Yang Yang等[19]通过宏基因组测序技术解析了中温大曲发酵过程中微生物群的分类和功能谱。目前还鲜有学者利用宏基因组测序技术对不同轮次的高温大曲群落进行比较研究,导致对高温大曲微生物群落的认识的短缺。因此本研究基于宏基因组学技术对3 个轮次(下沙、造沙、1 轮次)高温大曲群落进行精细解析,挖掘其群落结构与功能,并对不同轮次大曲间结构和功能差异进行分析,旨在为高温大曲的品质提升及生产优化提供理论依据。
1 材料与方法
1.1 材料与试剂
大曲样本于2021—2022年在贵州茅台酒厂采集,参考并优化麻颖垚等[23-24]采样方法,分别在下沙(XS)、造沙(ZS)、1轮次(YLC)取得25 个大曲样本,其中下沙轮次7 个(采样时间为10月5日—10月30日),造沙轮次9 个(采样时间为11月10日—12月10日),1轮次9 个(采样时间为12月25日—1月20日),装入无菌自封袋中。样品采集之后置于冰盒回实验室后迅速放入-80 ℃冰箱保存。
氢氧化钠、氯化钠、邻苯二甲酸氢钾、可溶性淀粉、己酸、无水乙醇、葡萄糖 上海阿拉丁试剂有限公司;E.Z.N.A.®Soil DNA Kit 美国Omega BioTek公司;琼脂糖 生工生物工程(上海)股份有限公司;FastPfuPolymerase 北京全式金生物技术有限公司;AxyPrep DNA Gel Extraction Kit 美国Axygen公司。
1.2 仪器与设备
NanoDropTM8000超微量分光光度计、VeritiPro聚合酶链式反应(polymerase chain reaction,PCR)仪美国Thermo Fisher Scientific公司;ExperionTM全自动电泳系统 美国Bio-Rad公司;高速台式冷冻离心机 湖南湘仪仪器有限公司;MGISEQ-2000测序仪 深圳华大智造公司。
1.3 方法
1.3.1 大曲样品DNA提取与检测
取2.0 g大曲样品置于50 mL离心管中,加入20 mL磷酸盐缓冲液(phosphate buffered saline,PBS),涡旋混匀10 min,然后2000 r/min、4 ℃离心5 min,将上清液转移至100 mL离心管中。收集到的上清液于12000 r/min、4 ℃离心10 min,沉淀用10 mL PBS洗涤一次,再2000 r/min、4 ℃离心10 min,收集沉淀,最后将收集的沉淀用1mL的PBS重悬备用。重悬的沉淀采用E.Z.N.A.®Soil DNA Kit进行提取,详细的实验步骤参照该试剂盒说明书,提取的DNA用50 μL RNA free water溶解,经1%琼脂糖凝胶电泳和分光光度计检测浓度及纯度。按照上述步骤重复提取大曲样品总DNA 3 次,将3 次提取的DNA样品混合并混匀,于-80 ℃保存备用。
1.3.2 宏基因组测序
文库构建:取1 μg大曲基因组DNA,使用超声波仪打断,打断后的样品用磁珠进行片段选择,使得样品条带集中在200~400 bp左右;修复双链cDNA末端,并在3’末端加上A碱基并对连接产物进行扩增,扩增产物用磁珠进行产物纯化回收;将回收后的扩增产物变性为单链,接着在环化反应体系中充分混匀适温反应一定时间,可得到单链环形产物,消化掉未被环化的线性DNA分子后,即得到最终的文库并进行检测。
DNBSEQ平台测序:检测合格文库安排上机测序(DNBSEQ),单链环状DNA分子通过滚环复制形成一个包含多个拷贝的DNA纳米球,接着将得到的DNB采用高密度DNA纳米芯片技术,加到芯片上的网状小孔内,通过联合探针锚定聚合技术进行测序(送深圳华大基因测序),测序数据已上传至NCBI(项目编号:PRJNA884532)。
1.3.3 大曲酶活力测定
采取二硝基水杨酸法[25],分别对大曲糖化酶与α-淀粉酶活力进行检测。
1.3.4 生物信息分析
测序数据质控:剔除含有10%不确定碱基(N碱基)的reads;剔除含有测序接头序列的reads(有15 个碱基或更长区域比对到接头序列);剔除含有50%以上低质量碱基(Q<20碱基)的reads。
序列拼接组装:使用Megahit拼接软件对Clean Data进行拼接组装,过滤掉300 bp以下的片段。
非冗余基因集的构建:将所有样品的预测基因序列用CD-HIT软件进行聚类,构建非冗余基因集。将样品所有的高质量reads与非冗余基因集进行比对,统计基因在对应样品中的丰度信息从而构建geneprofile。
基因预测:使用MetaGeneMark(http://topaz.gatech.edu/GeneMark/meta_gmhmmp.cgi)对拼接结果中的contig进行基因预测,并将其翻译为氨基酸序列。基因预测是指通过对组装的基因组序列进行分析,根据已知生物的基因结构知识或数据库序列识别其所包含的基因等功能区域;编码基因预测是识别基因组序列上所包含的蛋白质编码区域,通过在基因组序列上寻找开放阅读框实现。而高质量基因集Gene Catalogs则是在基因预测基础上进行去冗余操作得到。
物种稀疏性曲线分析:为验证本次大曲样本数量具有足够的物种覆盖程度,从样本中随机抽取一定数量的个体,统计出这些个体所代表物种数目,并以个体数与物种数构建曲线。它可以用来比较测序数量不同的样本物种的丰富度,也可以用来说明样本的取样大小是否合理。分析采用对样本进行随机抽样的方法,以抽到的样本的物种个数构建稀疏曲线,末端部分呈现上升的趋势表明抽样量不足,当曲线末端上升趋势趋于平缓时则表明采样量足够。
物种注释与分析:使用软件Kraken2[26]对宏基因组数据物种注释,获得物种的表达丰度,分析不同样品或者分组间的相似或差异性。宏基因组物种分析包括物种丰度的柱状图和热图分析、α多样性指数(Shannon指数)、β多样性分析(Bray-Curtis距离)和线性判别分析(linear discriminant analysis effect size,LEfSe)[19]。
基因功能注释与分析:对非冗余基因使用软件Diamond(https://github.com/bbuchfink/diamond)的BLASTP功能进行功能注释[27],主要注释数据库为京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)[28]及碳水化合物-活性酶(carbohydrate-active enzymes,CAZy)[29],并对注释结果进行功能差异分析,主要包括主坐标分析(principal co-ordinates analysis,PCoA)、Anosim组间(内)差异分析。
2 结果与分析
2.1 大曲宏基因组测序与组装质量分析
本次宏基因组测序共25 个样品,平均组装长度为100.5 Mb,共检测到的基因目录为438720 个。表1是对大曲样本测序数据进行组装分析以及基因预测和去冗余后的结果,显示随着轮次的进行,大曲样本的组装总长、基因数及去冗余基因数呈现递增趋势。

表1 大曲测序数据组装以及基因集结果统计Table 1 Statistics of sequencing data and gene assembly of microbial community in Daqu
本次宏基因测序样本基因片段长度集中在0~2000 nt 范围内,短片段长度基因占据大多数(图1a);Venn图显示各轮次样本基因种类具有较大差别(图1b);物种稀释曲线分析(图1c)表明3 个轮次的样本数量均具有足够物种覆盖度,说明其鉴定结果总体结果具备统计学意义,能够代表每个轮次的物种组成。

图1 大曲样本基因预测分析Fig.1 Gene prediction analysis of microbial community in Daqu
2.2 大曲样本多样性分析
通过Kraken2软件解析大曲在种水平上的物种结构组成,首次将多个轮次高温大曲微生物结构进行解析,共鉴定出5104 个物种。图2为大曲群落在属水平上的丰度前30的堆积图,可以发现大曲中主要以克罗彭斯特菌属(Kroppenstedtia)和芽孢杆菌属(Bacillus)为优势菌种。图3显示丰度前30的种,3 个轮次大曲种组成基本相同,但丰度稍有差异,大曲样本中优势菌种主要为K.eburnea和芽孢杆菌(B.sonorensis、B.velezensis、B.licheniformis),其中丰度最高的为K.eburnea,该菌种在芝麻香型白酒高温大曲中也有发现,在系统发育中属于高温放线菌科(Thermoactinomycetaceae),能够较好地适应大曲高温发酵[30],在本研究中发现其为酱香型高温大曲的主要菌种,且在样本中占比的相似度最高。

图2 大曲样本微生物群落在属水平上的组成Fig.2 Microbial composition at genus level in Daqu

图3 大曲样本微生物群落在种水平上的组成Fig.3 Microbial composition at species level in Daqu
通过计算Shannon指数得出各轮次α多样性指数(图4a),发现各轮次大曲样品内多样性水平基本一致。而根据丰度矩阵计算各轮次内样本间Bray-Curtis距离衡量β多样性(图4b),发现造沙多样性较其他2 个轮次高,推测该轮次作为下沙与1轮次之间的过渡所致,造沙阶段(11—12月)气候波动较大导致群落干扰程度增加,根据群落中度干扰理论[31],大曲中不同样本间物种多样性会更丰富。LEfSe(图5)可知,造沙差异微生物只有2 种,1轮次有17 种,说明造沙轮次与其余2 个轮次的群落物种差异较小,而1轮次与其余2 个轮次的群落差异较大,具有更多统计学差异的生物标识[13]。
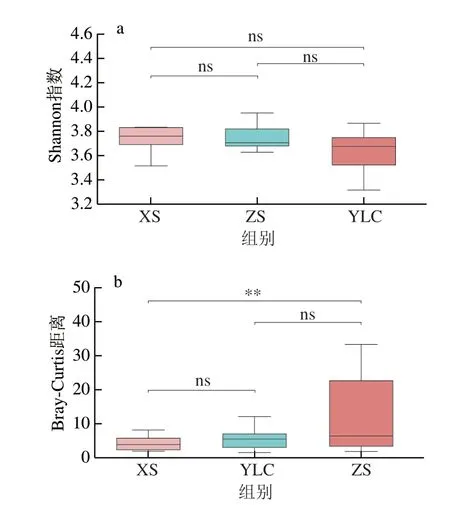
图4 大曲样本多样性Fig.4 Diversity of microbial community in Daqu samples

图5 不同轮次生产用曲的标记微生物Fig.5 Marker microorganisms in Daqu samples
2.3 大曲宏基因功能注释与分析
2.3.1 基因集KEGG注释结果
将大曲基因集注释到KEGG二级功能分类的19 条通路途径中,全局和概述图谱被注释到的基因数最多,其次是碳水化合物代谢与氨基酸代谢,三者均属于KEGG一级功能“代谢”分类下的二级代谢功能(图6)。全局和概述图谱的KEGG三级功能分类包括主要代谢通路,其次是碳代谢、翻译、单向转导、折叠、分选和降解以及其他次生代谢的生物合成途径,包含生物生存所必须的主要代谢功能或过程,因此在大部分研究中其分析结果基因数目最多[25]。大曲原料主要为小麦,其含有较为丰富的碳水化合物与蛋白质,因此碳水化合物代谢与氨基酸代谢相关基因也会比较丰富。

图6 KEGG二级分类注释图Fig.6 Secondary classification annotation in KEGG
2.3.2 基因集CAZy注释及淀粉水解相关酶活力测定
CAZy数据库中主要涵盖6 大功能类:糖苷水解酶(glycoside hydrolases,GHs),糖基转移酶(glycosyl transferases,GTs),多糖裂合酶(polysaccharide lyases,PLs),碳水化合物酯酶(carbohydrate esterases,CEs),辅助氧化还原酶(auxiliary activities,AAs)和碳水化合物结合模块(carbohydrate-binding modules,CBMs)。如图7a所示,在CAZy注释6大功能中,GHs注释结果最多,说明大曲在执行碳水化合物代谢功能中,GHs是促进多糖的水解为主要酶系。大曲中淀粉降解相关的关键酶主要为α-淀粉酶和糖化酶[32-33],酶活力检测结果显示(图7b),造沙大曲糖化酶和α-淀粉酶活力最高,下沙和造沙酶活力显著高于1轮次(P<0.05),造沙轮次GHs相关基因数目为2050,高于其余2 个轮次。
2.3.3 大曲基因功能差异分析
不同轮次大曲的主要功能基本一致(图8),但在不同轮次间存在差异。从图8a、d可以看出,大曲功能基因数组间组内差异基本一致,组间差异(蓝色柱子)显著大于下沙和1轮次组内差异,但与造沙相比无显著差异。在图8c、f显示出相似规律,下沙和1轮次在功能聚类上有显著差别,这可能是下沙轮次主要生产时间(10月份)与1轮次生产(1月份)处于温湿度差异比较大的2 个阶段所致。虽主要功能种类二者无异,但在功能基因数上还是有较为显著差异(图8b、e),造沙作为1轮次和下沙之间的过渡轮次,其组内与组间差异一致(图8a、d),在PCoA聚类分析上造沙轮次包含其余2 个轮次,而其余2 个轮次区分度较高,在聚类上进一步反映了造沙作为过渡轮次用曲的功能特点。
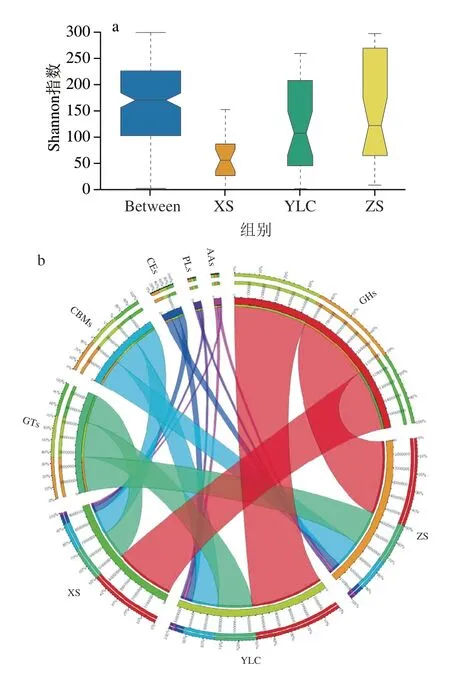
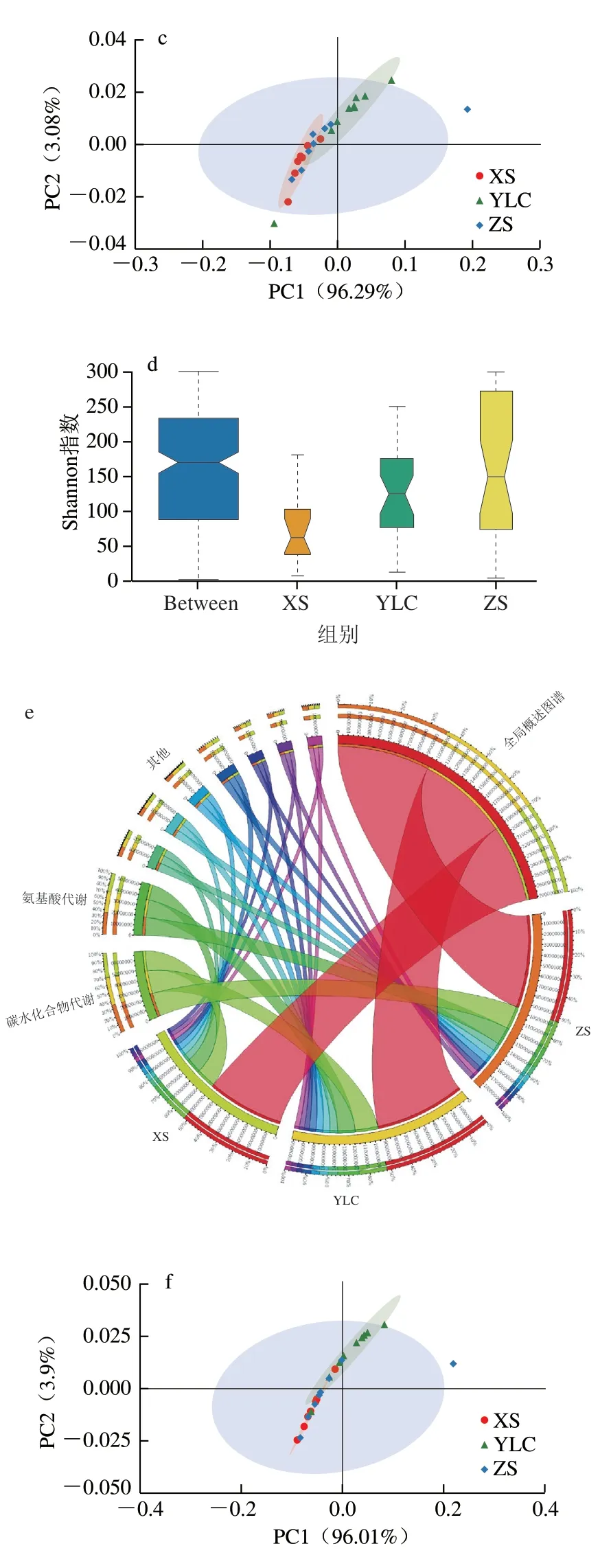
图8 功能差异分析Fig.8 Functional difference analysis
3 结论
本研究基于宏基因组技术分析高温大曲的微生物群落结构与功能,挖掘不同轮次生产用曲群落多样性信息与功能特征,得出如下主要结论:
1)本研究将宏基因组学技术应用于多轮次高温大曲微生物结构及其功能解析,补充高温大曲在种水平上的研究结果,明确高温大曲中以K.eburnea为优势菌种,多种芽孢杆属细菌为亚优势种共同构建酱香型大曲微生物群落;不同大曲物种α多样性水平基本一致,造沙β多样性略高于其余2 个轮次;1轮次差异物种最丰富,具有更多统计学差异的生物标识。
2)通过KEGG与CAZy 2 个通用数据库对大曲基因功能进行注释与分析,发现各轮次大曲主要执行代谢功能,碳水化合物代谢与氨基酸代谢为其中最主要的功能;淀粉水解相关酶系中,GHs被注释到的基因数最多;不同轮次大曲功能种类基本一致,但功能基因数目存在差异,其中下沙与1轮次大曲存在差异,造沙与其余2 个轮次无明显差异,可能由于造沙处于其余2 个轮次的过渡阶段所致。
3)本研究只采用了生产过程中前3 个轮次的大曲样本,发现不同轮次大曲微生物群落结构与功能组成基本一致,在实际使用过程中功能表现出现差异可能是受到生产环境干扰,该研究结果为后续大曲微生物资源保护、开发大曲判别标准及生产质控奠定了一定研究基础。后续研究中可进一步对优势菌株进行筛选应用,分析优势菌株在主要代谢通路中的作用,并进一步验证受到干扰条件下关键酶基因的应激表达情况,以期为大曲的品质提升、酒体品质的提升、微生物功能基因库的挖掘等方面提供更加系统的理论依据。
猜你喜欢
昆明医科大学学报(2022年2期)2022-03-29
食品安全导刊(2021年20期)2021-08-30
今日农业(2021年11期)2021-08-13
戏曲研究(2020年2期)2020-11-16
中华戏曲(2020年1期)2020-02-12
遵义(2017年1期)2017-07-12
遵义(2017年5期)2017-07-05
水生生物学报(2015年1期)2015-02-28
遗传(2014年3期)2014-02-28
世界科学(2014年8期)2014-02-28
