
Kniest发育不良合并脊柱后凸1例报道并文献复习
2023-09-07 00:42焦洋梁锦前陈唯韫沈建雄
中华骨与关节外科杂志 2023年7期
焦洋,梁锦前,陈唯韫,沈建雄
Kniest 发育不良(Kniest dysplasia, KD;OMIM:156550)是由COL2A1基因突变引起的骨骺发育异常疾病,呈常染色体显性遗传[1]。KD的发病率约1/1 000 000,国内外仅有少数病例报道[2-5]。KD 主要临床表现包括躯干和四肢短小、关节粗大、脊柱后凸、面部发育不良、严重近视和听力损失[6]。手术治疗KD 合并脊柱后凸的相关报道罕见。北京协和医院收治1例KD合并脊柱后凸患者,成功施行后路脊柱融合手术,并随访1年。现报道如下,同时进行相关文献复习。
1 临床资料
1.1 病例资料
患者,男,18 岁,因“发现脊柱后凸12 年,进行性加重”入院。患者足月顺产,1岁6个月时仍无法独立行走,伴有膝关节、肘关节肿大,于当地医院就诊,X线检查提示骨骺发育不良,基因检测提示COL2A1基因IVS18+1G>C 突变,考虑诊断KD,患者未接受治疗。12 年前患者家属发现患者出现脊柱后凸畸形,近年来脊柱后凸缓慢加重,患者无背痛、呼吸困难、四肢感觉异常、肌力下降、大小便异常等不适。既往高度近视、听力下降。家族中无类似疾病史。
本次就诊查体:身高102 cm,体重51 kg,无法行走。身材矮小、躯干短、颈短、圆脸、面部凹陷、眼球突出、钟形胸廓。双侧腕关节、肘关节、膝关节肿大。髋、膝关节活动严重受限:双侧髋关节屈曲100°,后伸-50°,外展20°,内收15°,内旋20°,外旋20°;双膝关节屈曲110°,伸直-75°。颈椎活动严重受限:前屈、后伸10°,左右侧屈25°,左右旋转45°;腰椎活动轻度受限:前屈45°,后伸15°,左右侧屈30°。脊柱胸段呈后凸畸形(图1)。四肢及躯干感觉对称正常,四肢肌张力正常但肌容积下降、四肢肌力Ⅴ级。双侧膝反射、跟腱反射未引出。双侧巴宾斯基征(-)。进一步完善影像学检查和相关实验室检查,全脊柱正侧位X线检查提示:椎体广泛扁平化,胸椎椎体前缘楔形变,胸段后凸伴矢状面失衡,T5-T12 Cobb 角96°,T10-L2 Cobb 角26°,矢状面轴向距离10.8 cm;股骨干骺端扩大,呈“哑铃状”,股骨颈宽短,双侧髋臼宽大且发育不良。脊柱CT 重建提示:脊柱椎体高度减低,椎体骨骺发育不良,椎间隙不规则变窄(图2)。脊柱MRI提示颈椎、胸椎、腰椎椎管狭窄,无硬膜囊受压表现。骨代谢实验室检查结果显示血钙、血磷、维生素D 及碱性磷酸酶水平均正常。
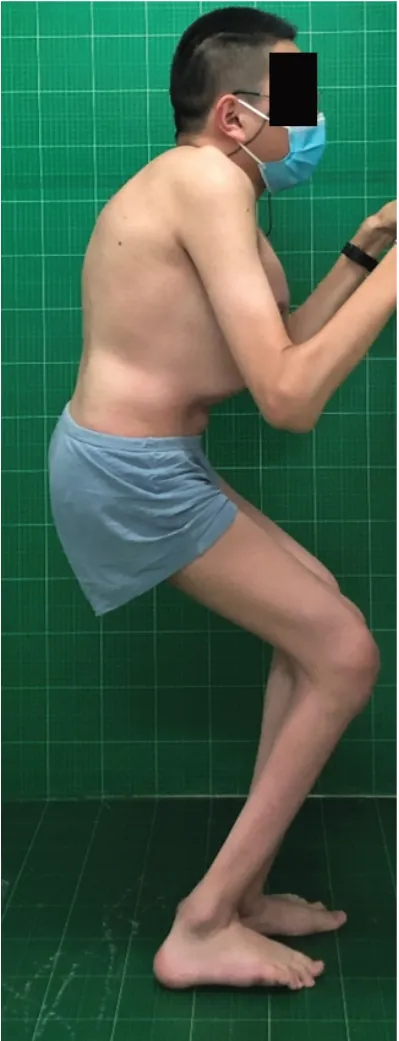
图1 患者术前大体像,可见鸡胸,膝关节、髋关节伸直受限
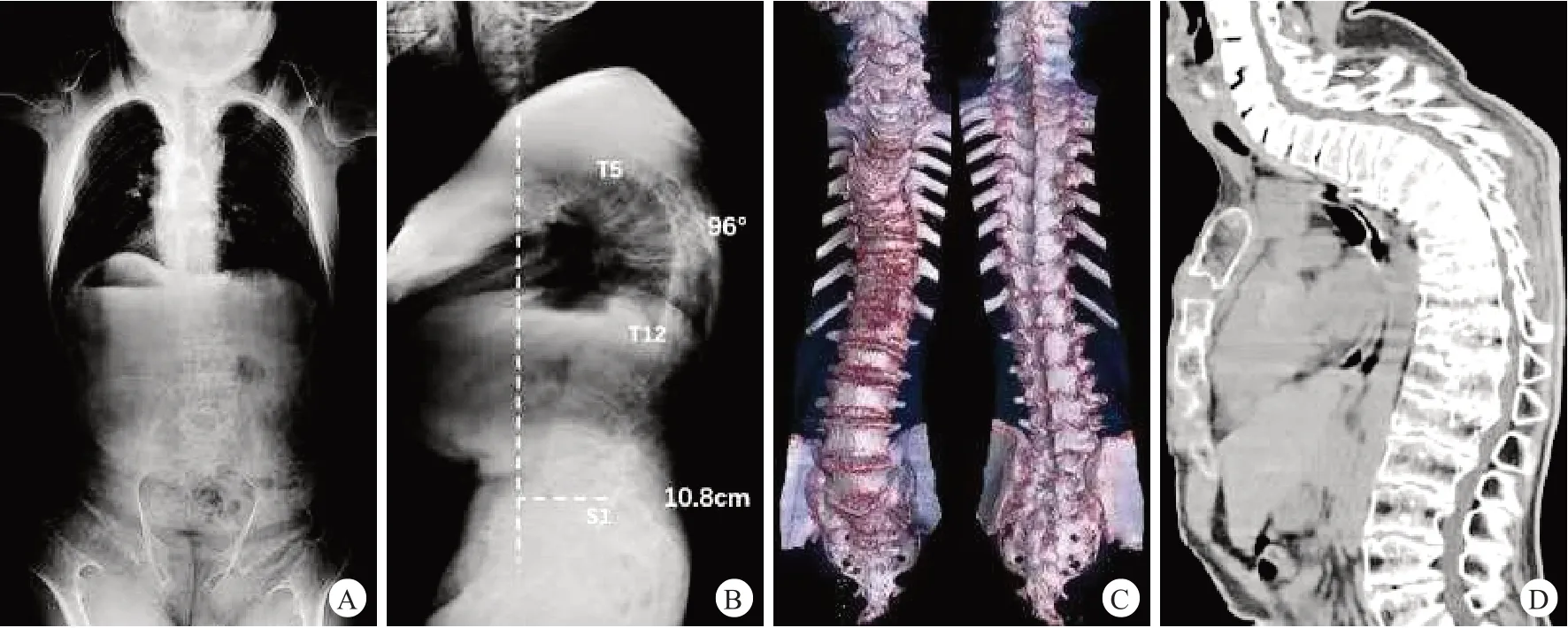
图2 患者术前影像学表现
1.2 诊疗经过
本例患者的临床表现为早发性四肢关节粗大伴活动受限,躯干缩短伴鸡胸,视力、听力异常,脊柱后凸。X线检查可见股骨干骺端呈“哑铃状”改变、脊柱椎体广泛骨骺发育不良,骨代谢实验室指标均正常。基因检测提示COL2A1基因IVS18+1G>C突变。结合患者的临床表现、影像学资料及基因检测结果,诊断为KD[7]。
考虑到患者目前脊柱后凸角度已达96°,近年来逐渐加重。并且患者存在发育性的胸椎管狭窄,若后凸持续加重,很可能压迫脊髓而引起神经损伤。此外,患者目前的矢状面轴向距离为10.8 cm,呈矢状面失衡状态。为了改善躯体的平衡并及时控制患者脊柱后凸畸形进展,对其进行了全身麻醉下脊柱后路融合、内固定、植骨融合术,范围为T2-T12。麻醉时,由于颈椎后伸严重受限,气管插管难度大,最终通过可视喉镜辅助成功完成气管插管。手术时,在T3-T11 范围进行SP 截骨(Smith-Petersen osteotomy,SPO),在T3-T12 间置入椎弓根螺钉并使用钴铬钼合金棒完成连接。自后凸的顶椎T8 向头、尾端椎体逐个进行适度加压,改善胸段后凸,透视满意后,锁紧椎弓根螺钉螺母。使用颗粒化的自体松质骨及同种异体骨行Moe 植骨。术中出血约550 mL,手术时间2.5 h。术中脊髓神经监测未见异常信号。术后患者顺利拔管。术后1年复查伤口愈合良好,全脊柱正侧位X线检查及大体像提示脊柱内固定位置良好,脊柱后凸未见继续加重,胸后凸Cobb 角改善至78°,矢状面轴向距离由10.8 cm矫至7.2 cm(图3)。
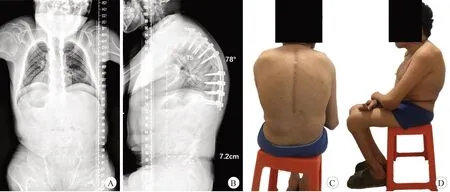
图3 患者术后1年影像学表现及大体像
2 讨论
KD由Kniest在1952年首次描述而命名,是一种罕见的常染色体显性遗传骨骺发育异常疾病[5,8]。位于染色体12q13.11区的COL2A1基因是其致病基因,该基因的突变可导致维持骨关节完整性的主要蛋白——Ⅱ型胶原蛋白发生改变,导致多种临床表现和程度不同的Ⅱ型胶原病,包括脊柱干骺端发育不良、先天性脊柱骨骺发育不良、Stickler 发育不良、2 型软骨发育不良等十余种疾病[9-11]。KD与其他Ⅱ型胶原病的鉴别需要进行基因检测,除了COL2A1基因IVS18+1G>C突变外,KD其他基因突变位点包括c.905C>T p.(Ala302Val)、c.1366G>C p.(Gly456Arg)、c.1023+1G>T、c.1681-1G>C、c.3383G>T p.(Gly1128Val)和c.1448G>A p.(Gly483Glu)[12-13]。尽管基因检测是诊断KD的利器,但KD的明确诊断首先基于详尽的病史采集和体格检查,躯干和四肢短小、关节粗大、关节活动受限、脊柱后凸、面部发育不良、严重近视和听力损失均有重要临床诊断意义[14]。除Ⅱ型胶原病外,由于KD患者具有鸡胸、身材矮小、关节肿大的特征,因此还需与佝偻病和黏多糖贮积症进行鉴别:KD患者骨代谢实验室指标均正常,可与佝偻病鉴别;KD患者智力多正常,且长骨X线检查可见骨骺发育不良,可与黏多糖贮积症相鉴别[15-16]。
尽管KD 会严重影响患者的运动能力、听力及视力,但目前尚无针对其发病机制的治疗方法,但通过针对性的外科手术,可以改善患者的生活质量。Merrill等[17]报道了1例KD患者合并寰枕区不稳定,通过枕骨至C2的固定融合手术,寰枕区不稳定得到了有效的治疗。Sitoula等[18]报道了1 例KD 患者合并寰枢椎不稳定,通过在寰枢椎使用钛缆固定、自体髂骨植骨融合,实现了骨性融合。Buschang等[19]应用微型螺钉种植体对KD 患者合并的颌面畸形进行了满意的矫正。Husain等[20]对一例KD患者进行了人工耳蜗植入、下颌牵张术、腭成形术和喉气管重建,使患者的听力及吞咽、呼吸功能得到一定改善。但目前已发表的关于KD的病例中未见合并脊柱后凸的手术治疗的报道,这可能与疾病的罕见性和对疾病的认识有限有关。
目前尚无文献总结KD 合并脊柱后凸的手术指征,但作者认为,当胸后凸超过70°并呈进行性加重趋势、躯干矢状面失衡伴有明显背痛、脊柱后凸严重影响生活质量时,应考虑手术治疗。在脊柱后凸矫形时,KD患者面临不同级别截骨的选择、牵引后再进行后路手术或前后路联合手术等选择。但由于KD患者颈椎及多处关节的活动度严重受限,所以脊柱后凸矫形的程度需要与其颈椎的代偿能力相匹配。本例患者髋关节、膝关节术前均处于屈曲畸形状态,活动范围小(髋关节屈伸活动范围50°,膝关节活动范围45°),而颈椎前屈活动度仅10°,对后凸矫形的代偿能力差。若胸段后凸矫形过多,术后髋、膝关节需增加屈曲程度以维持正常视野,这会进一步增加脊柱矢状面的失衡。为了提高患者的生活质量,作者所在团队最终根据颈椎前屈的活动度确定了胸段后凸的矫正范围,选择对后凸进行10°~30°的适度矫形。确定矫形范围后,由于KD 患者的脊柱椎体存在广泛的骨骺发育不良,椎间隙窄,这使得患者进行Schwab高级别截骨手术(如经椎弓根截骨或全椎体切除)时易损伤神经并发生假关节。Lenke等[21]报道使用全椎体切除术治疗重度僵硬性脊柱侧后凸畸形时,虽然矫形率达67%,但神经损伤相关并发症的发生率达27%。Mallepally等[22]报道使用全椎体切除术治疗重度脊柱后凸时,内固定失败、假关节的发生率显著升高。因此,作者所在团队选择一期后路多节段SPO 截骨,适度矫形脊柱后凸。另外,为避免假关节相关风险,术中使用强度更高的钴铬钼合金棒进行矫形,并在Moe植骨时选用颗粒化的自体松质骨结合一个同种异体的冰冻股骨头。通过低级别截骨矫形,加强内固定强度及大量、充分的植骨来避免假关节的形成。本例患者术后随访1年未诉背痛等不适,复查全脊柱正侧位X 线检查提示胸后凸Cobb 角由术前96°矫至78°,矢状面轴向距离由10.8 cm 改善至7.2 cm,但远期情况仍需密切随访。
综上,KD 作为一种罕见的遗传性骨骺发育不良疾病,基因检测有利于明确诊断。目前尚无根治KD的治疗方式,对于KD 合并的骨骼畸形如寰枢椎区不稳定及脊柱后凸,针对性的外科手术可以提高患者的生活质量。根据本病例的治疗经过与文献复习,KD 合并脊柱后凸患者进行脊柱后路固定融合手术时,后凸矫形的程度需与颈椎代偿能力匹配。
【利益冲突】所有作者均声明不存在利益冲突
猜你喜欢
南方农机(2022年7期)2023-01-04
保健医苑(2022年1期)2022-08-30
保健医苑(2022年6期)2022-07-08
放射学实践(2021年5期)2021-05-21
颈腰痛杂志(2020年1期)2020-03-27
脊柱外科杂志(2020年1期)2020-02-12
中国生殖健康(2019年10期)2019-01-07
华人时刊(2017年19期)2017-11-21
现代商贸工业(2016年34期)2016-03-13
焊接(2016年5期)2016-02-27
