
密度泛函理论与分子力学方法研究沥青质分子间的π-π堆积作用
2023-09-05 00:33:56罗辉吕慧栋周嘉安琪邓文安李传南国枝
中国石油大学学报(自然科学版) 2023年1期
罗辉 吕慧栋 周嘉安琪 邓文安 李传 南国枝

摘要:為准确描述沥青质分子间的π-π堆积作用,采用密度泛函理论(DFT)和分子力学(MM)方法研究π-π堆积的势能面、质心距离(Re)和相互作用能(ΔEe)。结果表明,DFT-D、DFT/mGGA、MM以及采用DNP+基组的DFT/GGA方法在描述苯和沥青质的S型、T型和PD型二聚体时均可得到典型的势能面;DFT/mGGA-M06L方法的精确度最高,得到的Re和ΔEe与苯的参考值相比,误差均小于4.60%;S型沥青质二聚体的Re与苯的非常接近,均为0.388 nm,但ΔEe比苯的大十余倍;T型沥青质二聚体中近距离作用的原子数越多,ΔEe越大,但近距离作用的原子数只有一个或几个,导致ΔEe明显小于S型;PD型沥青质二聚体的ΔEe大于S型,且质心垂直距离(RV)在约0.35 nm时ΔEe最大(接近S型的2倍),此时二聚体的片层间距明显小于S型,结构最稳定。
关键词:沥青质聚集体;π-π堆积作用;密度泛函理论;分子力场
中图分类号:TE 622.1 文献标志码:A
引用格式:罗辉,吕慧栋,周嘉安琪,等.密度泛函理论与分子力学方法研究沥青质分子间的π-π堆积作用[J].中国石油大学学报(自然科学版),2023,47(1):189-196.
LUO Hui, L? Huidong, ZHOU Jiaanqi, et al. Study on π-π stacking between asphaltene molecules by DFT and MM methods[J].Journal of China University of Petroleum(Edition of Natural Science),2023,47(1):189-196.
Study on π-π stacking between asphaltene molecules by DFT and MM methods
LUO Hui, L? Huidong, ZHOU Jiaanqi, DENG Wenan, LI Chuan, NAN Guozhi
(State Key Laboratory of Heavy Oil Processing in China University of Petroleum (East China), Qingdao 266580, China)
Abstract:To gain insight about the π-π stacking in asphaltene molecular aggregates, density functional theory (DFT) and molecular mechanics (MM) methods were adopted to study the potential energy surface (PES), the equilibrium center of mass distance (Re) and the equilibrium interaction energy (ΔEe). The results show that the typical PES are obtained to describe the S-type (sandwich), T-type (T-shaped) and PD-type (parallel-displaced) dimers of benzene or asphaltenes by using DFT-D, DFT/mGGA, MM, or DFT/GGA with DNP+ basis set, where the DFT/mGGA-M06L method has the highest accuracy. The ΔEeand Recalculated by mGGA-M06L are consistent with those reported in the literature for benzene dimers, and the errors are less than 4.60%. The Reof the S-type asphaltene dimer is very close to that of benzene, both are 0.388 nm, and ΔEeis more than ten times larger than that of benzene. For the T-type asphaltene dimers, the more atoms in the short-distance, the greater the ΔEe, but there is only one or several atoms in the short-distance, resulting in the ΔEesignificantly smaller than that of the S-type dimer. The ΔEeof the PD-type asphaltene dimers are greater than that of the S-type, and the ΔEeis the largest when the vertical distance (RV) is about 0.35 nm, which is close to twice that of the S-type. In this case, the interlamellar spacing of the dimer is significantly smaller than that of the S-type, resulting in the most stable structural conformation.
Keywords: asphaltene aggregates; π-π stacking; density functional theory(DFT); molecular force field
沥青质作为石油中相对分子质量最大和分子结构最复杂的组分,具有低氢碳比(H/C)、高极性和高芳香性等特性,容易形成超分子聚集体而析出,导致石油在开采、储运与加工过程出现沉淀、堵塞、结焦等一系列技术问题[1-2]。深入认识沥青质的超分子聚集体及其聚集作用力,并据此破坏沥青质的超分子聚集结构,防止其聚集析出,对于石油的开采、储运与加工均具有重要的意义。石油沥青质一般具有较大的芳香片结构和S、N等杂原子[3],这种较大芳香片之间会形成π-π堆积片层结构,而杂原子之间会形成氢键,进而组装成超分子聚集体[4-9]。也就是说π-π堆积作用力和氢键作用力是石油沥青质超分子结构形成并稳定存在的重要推动力。当然由于石油沥青质分子结构的复杂性,形成超分子聚集体的推动力究竟是由哪种作用力主导的目前还存在一定的争议。π-π堆积作用是指芳香性体系中由于π电子的共轭分布而导致的分子间相互作用,是芳环结构中常见的相互作用。沥青质分子中含有若干个芳环,它们之间的π-π作用被认为是形成沥青质聚集体的关键作用之一[4-6],但是对π-π堆积的本质还没有统一的认识。WANG等[4]认为石油沥青质分子芳香片层间的π-π堆积作用的主要来源为色散力,任强等[6]发现除色散力可能还有少量的电子转移;蔡新恒等[7]认为沥青质分子间π-π作用的实质并不限于色散力,可以是分子间π电子云高度重叠及静电吸引;甚至在单体的电子给体和受体间还存在强烈的轨道相互作用[10]。得到的π-π堆积作用力大小也是千差万别。对于π-π堆积作用这种明显低于共价键、离子键等化学键的分子间弱相互作用力,目前仍然没有足够的实验手段来进行研究,而理论计算和分子模拟则可以方便地描述这些问题,尤其是密度泛函理论(DFT)和分子力学(MM)模拟是常用的也是较为经济的方法[11-12]。DFT方法在描述色散力等弱相互作用力上还存在一些不足[13-14]。MM方法形式更为简单,基于分子力场描述分子间的作用力,不考虑电子的作用,结果的准确性依赖于经验性的分子力場,在不同体系中的表现参差不齐。笔者为全面、准确地描述沥青质分子间的π-π堆积作用,对比分析DFT方法中不同的泛函以及MM方法中不同的分子力场在描述π-π堆积作用上的表现,在此基础上探究沥青质分子π-π堆积作用的类型、强度及其作用范围。
1 研究方法
1.1 沥青质模型分子
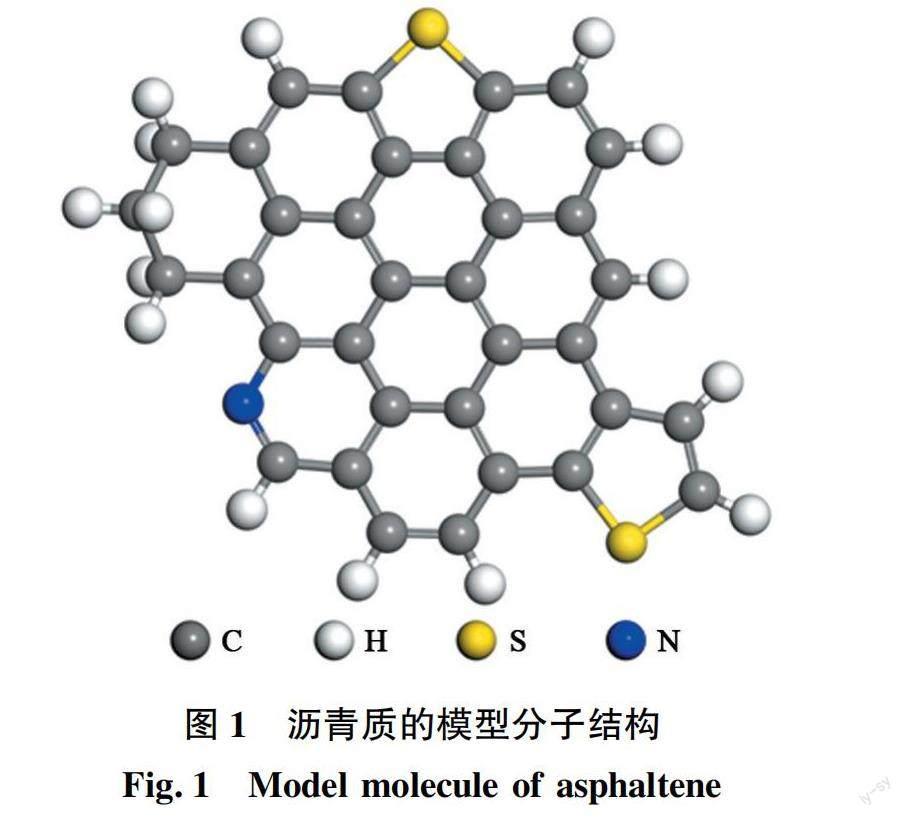
综合利用红外光谱、核磁共振(NMR)、X射线光电子能谱(XPS)等表征方法可获取沥青质分子的结构片段,如碳氢骨架结构片段和S、N、O等杂原子官能团信息,再结合沥青质的元素组成和相对分子质量,可构建出沥青质的平均模型分子。这种模型分子也是表述沥青质结构特性的有效方式之一。根据文献中典型沥青质的结构参数和杂原子官能团的分析结果,构建出沥青质分子的模型化合物结构[6]见图1。
1.2 DFT计算方法
沥青质超分子聚集体π-π堆积作用的DFT计算均在Material studio软件包的DMol3程序[15-16]中完成。采用Visualizer模块构建沥青质分子的初始构型,在DMol3程序中采用BFGS算法对初始构型进行几何全优化计算,结构优化的收敛判据为能量、最大力和最大位移,收敛精度分别为2.6255×10-2kJ/mol、5.251 kJ/(mol·?)和0.005 ?。自洽场(SCF)的收敛阈值为2.6255×10-3kJ/mol。结构优化完成后进行频率分析,确保所得的优化结构均为势能面上的极小点(无虚频)。
参照苯分子间的π-π堆积作用,构建平行(S型)、垂直(T型)和滑移平行(PD型)的二聚体构型(图2,其中RV为质心垂直距离)),采用不同的泛函计算二聚体的能量,分子之间的π-π堆积相互作用能(ΔE)为
ΔE= Eagg-2Emo.(1)
式中,Emo为单个沥青质分子单独存在时的能量,kJ/mol;Eagg为沥青质分子二聚体的能量,kJ/mol。
改变不同构型二聚体的质心距离(R),并计算其相互作用能ΔE,ΔE与R之间的关系曲线则为相互作用能的势能面。势能面曲线上最小值(或极小值)点的相互作用能和质心距离分别记为ΔEe和Re,即为该构型二聚体的π-π堆积作用强度和堆积作用距离。
采用的泛函包括广义梯度近似(GGA)中的PW91、PBE、BLYP等,以及杂化泛函B3LYP和mGGA中的M06-L、M11-L、TPSS等;同时对比是否对色散力等弱相互作用力进行校正(DFT-D);基组主要对比考察全电子双数字加极化基组(DNP)和引入了色散函数的DNP+基组。
1.3 MM计算方法
π-π堆积作用的MM模拟均在Material studio软件包的Forcite程序中进行。沥青质分子结构优化采用Smart方法,范德华和库仑相互作用的计算采用Atom based算法,截断半径为18.5 ?。结构优化的收敛判据为能量、最大力和最大位移,收敛精度分别为8.37×10-5kJ/mol,4.18×10-3kJ/(mol·?)和1.0×10-5 ?。
分子间π-π堆积作用能的计算采用的分子力场包括COMPASS、Dreiding、Universal、cvff、pcff等。
2 结果分析
2.1 DFT或MM方法对苯分子π-π堆積的影响
沥青质分子间的相互作用力包括氢键、π-π堆积、静电和范德华力等多种类型。采用从头算方法或DFT方法可以很好地描述其中的氢键和静电作用,但π-π堆积作用和范德华力,尤其前者与电子的瞬时动态密切相关,具有非局域性,对其进行准确描述的难度较大[17-18]。
高精度的从头算方法理论上可以计算得到足够精确的分子间相互作用力,但沥青质分子较大,包含的电子数多,DFT方法是唯一可接受的第一性原理方法。在确定DFT方法的可靠性前需要进行测试和验证。苯-苯二聚体分子间的π-π堆积作用最为简单也最为经典,以苯-苯二聚体的S型、T型和PD型结构模型为对象,测试几种流行的DFT泛函和MM分子力场描述π-π堆积作用的准确性。
采用DFT/GGA和杂化泛函B3LYP方法描述的苯二聚体分子间的相互作用势能面见图3(a)。典型的相互作用势能面随粒子间的距离分为近程、中程和远程3个区域:近程区以排斥力为主,其相互作用能ΔE>0,且随距离的增大,排斥力迅速下降;中程区以吸引力为主,其相互作用能ΔE<0,且随着距离的增大,吸引力占的比例越来越大,最终在距离Re处达到平衡而形成势能面的极小值ΔEe;远程区的排斥力可以忽略,只存在部分吸引力,且随着距离的增大,吸引力也逐渐减小,直至ΔE→0。由图3(a)可知,GGA下的PBE、PW91和BLYP,以及杂化泛函B3LYP采用DNP基组时,得到的S型苯二聚体的ΔE均大于0,其势能面并不是典型的势能曲面,因此这些方法明显不能用来描述π-π堆积作用。采用DNP+基组时,这些泛函得到的势能面均为典型的势能曲面,均有势能面极小值点,但与参考数据(ΔEe= -7.12 kJ/mol,Re= 0.388 nm)[19]对比,GGA-BLYP-DNP+和B3LYP-DNP+得到的ΔEe偏小很多、且Re偏大很多,GGA-PBE-DNP+和GGA-PW91-DNP+得到的ΔEe略有偏小,但Re偏大较多,因此采用DNP+基组时,这些泛函描述π-π堆积作用也不够准确。
图3(b)为引入色散力校正的DFT-D方法和引入动能密度及密度二阶梯度的mGGA方法描述的S型苯二聚体的势能面,得到的这些势能面均为典型的势能曲面,均有势能面极小值点。这些方法得到的Re均约为0.38 nm,与参考数据吻合。DFT-D/GGA-PW91与mGGA-M11L方法得到的ΔEe偏大很多,高估超过5 kJ/mol,DFT-D/GGA-PBE与DFT-D/mGGA-TPSS得到的ΔEe也偏大较多,高估超过1.3 kJ/mol,而DFT-D/GGA-BLYP、DFT-D/B3LYP和mGGA-M06L得到的ΔEe较为准确,仅高估约0.2 kJ/mol,误差不超过3%。因此DFT-D/GGA-BLYP、DFT-D/B3LYP和mGGA-M06L可较好地描述S型苯二聚体的π-π堆积作用。
MM方法是基于经典力学的计算方法,不考虑电子的运动、形式简单、计算速度快,但其经验势能函数中显含弱相互作用的相关项,因此也有可能用来描述π-π堆积作用。MM方法描述的S型苯二聚体的势能面如图3(c)所示。可以看出,MM方法得到的势能面均为典型的势能曲面,均有势能面极小值点。CVFF、Uinversal和Dreiding力场得到的ΔEe明显高估很多,不能准确描述π-π堆积作用;而Compass和PCFF力场得到的Re约为0.38 nm,与参考数据吻合,得到的ΔEe低估约为1.5 kJ/mol,精度不如上述的DFT-D和mGGA方法。
采用DFT/GGA、DFT-D、DFT/mGGA和MM等方法得到的T型和PD型苯二聚体的势能面如图4所示。由图4(a)看出,与S型类似,DFT-D、DFT/mGGA、MM、以及采用DNP+基组的DFT/GGA方法描述的势能面均为典型的势能曲面。与参考数据(ΔEe=-11.34 kJ/mol,Re=0.496 nm)[19]对比,mGGA-M06L的结果精确度高,得到的Re= 0.489 nm、ΔEe仅低估0.45 kJ/mol,误差为3.97%。DFT/D方法得到的Re均约为0.48 nm,与参考数据相差较小,但ΔEe高估很多,DFT-D/GGA-PBE、DFT-D/B3LYP和DFT-D/GGA-BLYP分别高估4.47、4.57和8.44 kJ/mol。采用DNP+基组的DFT/GGA-PBE-DNP+和DFT/GGA-PW91-DNP+得到的Re约为0.51 nm,ΔEe分别高估1.02和1.82 kJ/mol。分子力学方法MM/Compass和MM/PCFF得到的Re约为0.52 nm,ΔEe分别高估2.94和3.30 kJ/mol。
由图4(b)PD型(RV=0.36 nm)苯二聚体势能面看出,与参考数据(ΔEe=-11.09 kJ/mol,Re=0.166 nm)[19]对比,mGGA-M06L的结果同样是各方法中最精确的,得到的Re=0.166 nm、ΔEe高估0.51 kJ/mol,误差为4.60%。DFT/D方法得到的Re约为0.17 nm,ΔEe有所高估,DFT-D/B3LYP、DFT-D/GGA-PBE和DFT-D/GGA-BLYP分别高估2.08、2.28和2.50 kJ/mol。DFT/GGA- PW91-DNP+和DFT/GGA-PBE-DNP+得到的Re為0.18 nm,ΔEe分别低估0.27和1.25 kJ/mol。MM/Compass和MM/PCFF得到的Re约为0.28 nm,与参考数据相差较大,ΔEe分别高估2.67和2.77 kJ/mol。
2.2 沥青质分子的S型π-π堆积
参照S型苯二聚体的方法,采用DFT/GGA、DFT-D、DFT/mGGA和MM等方法计算S型沥青质二聚体的势能面,结果见图5和表1。与S型苯二聚体类似,DFT/GGA采用DNP基组时得不到典型的势能曲面,无法描述沥青质分子的π-π堆积作用。DFT-D、DFT/mGGA、MM、以及采用DNP+基组的DFT/GGA方法均可得到典型的势能曲面,但各势能面的Re和ΔEe各不相同。
从描述苯二聚体S型、T型和PD型的π-π堆积作用结果来看,mGGA-M06L方法的精确度最高,其计算的Re和ΔEe与参考值相比,误差均小于4.60%。因此将mGGA-M06L方法计算的结果作为参考值,质心距离Re=0.388 nm、最大相互作用能ΔEe=-78.50 kJ/mol。与S型苯二聚体的π-π堆积作用(ΔEe=-7.12 kJ/mol,Re= 0.388 nm)相比,沥青质二聚体的Re与苯的非常接近,但ΔEe大了十余倍。这可以说明,虽然沥青质分子的结构更为复杂,但其S型π-π堆积作用的本质与苯是一致的,只是沥青质分子更大,从而ΔEe更大。
对比各方法的结果,DFT-D方法计算的Re在0.38~0.39 nm,与参考值接近,特别是DFT-D/GGA-PBE计算的ΔEe为-80.30 kJ/mol,仅比mGGA-M06L的结果(参考值)高估1.80 kJ/mol,表现较好。采用DNP+基组的DFT/GGA方法低估Re并高估ΔEe,而MM方法不同程度地高估Re和高估ΔEe。
从S型堆积的构型(图2)来看,由于沥青质的芳香核中含有环烷环,有部分H原子不在芳香核的平面上,按S型堆积时会存在一定的斥力,导致堆积结构不紧凑、不稳定。为消除这些因素的影响,将S型堆积上层的沥青质分子绕质心旋转一定的角度,使这些不在平面的原子错开。采用mGGA-M06L方法计算不同旋转角度的S型沥青质二聚体的
Re和ΔEe,结果见图6。
由图6可知,S型堆积的沥青质分子旋转错开时,二聚体的Re减小、同时ΔEe增大,这说明不在芳香核平面的原子错开后再进行S型堆积,可使堆积结构更为紧凑和稳定。但错开到一定角度时,如大于60°后再继续旋转,此时不在芳香核平面的原子已完全错开,二聚体的Re和ΔEe不再有明显的变化。
2.3 沥青质分子的T型π-π堆积
采用DFT-D、DFT/mGGA和MM等方法计算得到T型沥青质二聚体的势能面,如图7所示,计算得到的Re和ΔEe见表2。同样以mGGA-M06L的结果为参考值,质心距离Re=0.750 nm、最大相互作用能ΔEe=-32.06 kJ/mol。与mGGA-M06L相比,其他方法得到的ΔEe均有所高估。DFT-D/GGA-PBE、DFT/GGA-PBE-DNP+和DFT/GGA-PW91-DNP+计算的Re约为0.75 nm,与参考值接近;MM方法计算的Re有所高估,而DFT-D/GGA-BLYP计算的Re则有所低估。相较而言,DFT-D/GGA-PBE的表现相对较好,其Re=0.750 nm、ΔEe比参考值高估11.80 kJ/mol。
比较苯的二聚体结构可知,其T型结构(ΔEe参考值为-11.34 kJ/mol)的稳定性高于S型结构(ΔEe参考值为-7.12 kJ/mol)。但对于沥青质二聚体而言,各方法计算得到的T型二聚体的ΔEe均明显小于S型的,说明T型的沥青质二聚体不如S型稳定,这与苯二聚体的情况正好相反。
沥青质的分子结构比苯复杂,其T型二聚体可能存在多种不同的结构,采用mGGA-M06L方法计算了不同结构的T型沥青质二聚体的Re和ΔEe,结果见图8。由图8看出,各T型沥青质二聚体的ΔEe均较小,均明显小于S型的,即这些T型沥青质二聚体均不如S型稳定。比较不同结构的T型二聚体可以发现,垂直方向与水平方向上的分子近距离作用的原子数越多,其ΔEe越大,特别是当垂直方向的分子只有一个H原子与水平方向分子近距离作用时,ΔEe仅稍大于苯二聚体的。这是由于π-π堆积作用的本质属于典型的色散力[6, 20-21],近距离作用的原子数越多,色散力越大,从而π-π堆积作用越大。虽然沥青质的分子较大,但按T型结构进行堆积时,近距离作用的原子数较少,只有一个或几个,导致堆积作用明显小于S型的。
2.4 沥青质分子的PD型π-π堆积
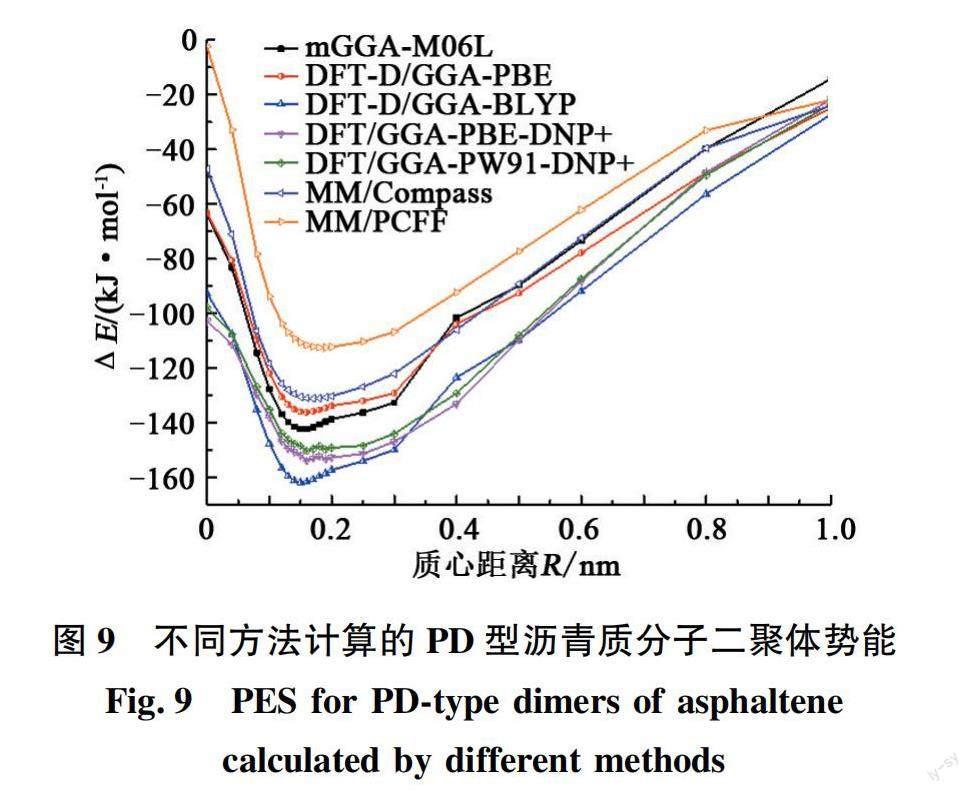
图9为各方法计算得到的PD型(质心垂直距离RV=0.36 nm)沥青质二聚体的势能面,对应的Re和ΔEe见表3,参考值mGGA-M06L方法得到的质心水平距离Re= 0.156 nm、ΔEe= -142.36 kJ/mol。对比各方法的结果可知,除了MM/PCFF得到的Re有所高估之外,其余各方法计算得到的Re均与参考值比较接近,在0.15~0.17 nm之间。DFT-D/GGA-PBE、MM/Compass和MM/PCFF得到的ΔEe有所低估,而DFT/GGA-PW91-DNP+、DFT/GGA-PBE-DNP+和DFT-D/GGA-BLYP计算的ΔEe则有所高估。相较而言,DFT-D/GGA-PBE的表现相对较好,其Re= 0.160 nm,ΔEe比参考值低估6.22 kJ/mol。
苯二聚体的PD型结构的ΔEe(RV=0.36 nm时参考值为-11.09 kJ/mol)与T型(参考值为-11.34 kJ/mol)接近,高于S型(参考值为-7.12 kJ/mol),即苯二聚体的PD结构比S型稳定,这也是通过实验证实过的[22]。对于沥青质二聚体,各方法计算得到的PD型二聚体的ΔEe均明显大于S型的,此外与旋转错开的S型二聚体(图6)相比,PD型二聚体的ΔEe也明显更大,这就说明PD型的沥青质二聚体比S型的更为稳定,与苯二聚體的规律一致。
采用mGGA-M06L方法比较了质心垂直距离RV不同的条件下PD型二聚体的Re和ΔEe,数据如表4所示。表4表明,RV不同时,PD型二聚体的Re
均比较接近,但ΔEe明显不同。当RV较小时,RV增大导致ΔEe增大,但RV较大时,则RV增大导致ΔEe减小,也就是说,RV有最佳值,此时PD型二聚体的ΔEe最大,即最稳定。RV的最佳值约为0.35 nm,明显小于S型沥青质二聚体的质心距离Re(参考值为0.388 nm),此时的ΔEe为-146.50 kJ/mol,接近S型(-78.50 kJ/mol)的2倍。PD型二聚体的片层间距明显小于S型的,比S型更为紧凑,也更为稳定。
3 结 论
(1)DFT和MM方法在描述苯π-π堆积作用时的表现表明,DFT-D、DFT/mGGA、MM以及采用DNP+基组的DFT/GGA方法均可得到典型的势能面,其中DFT/mGGA-M06L方法的精确度最高,计算的S型、T型和PD型苯二聚体的Re和ΔEe与参考值相比,误差均小于4.60%。
(2)DFT-D、DFT/mGGA、MM以及采用DNP+基组的DFT/GGA方法在描述沥青质的S型、T型和PD型苯二聚体时均可得到典型的势能面。以DFT/ mGGA-M06L的结果为参考值,对于S型和T型,其余的方法均有所高估ΔEe,DFT-D方法计算的Re接近参考值;对于PD型,除MM/PCFF外各方法计算的Re均接近参考值,DFT-D/GGA-BLYP和采用DNP+基组的DFT/GGA方法高估ΔEe,而DFT-D/GGA-PBE和MM方法则低估ΔEe;总体而言,DFT-D/GGA-PBE的表现相对较好。
(3)对于S型沥青质二聚体,其Re与苯的非常接近,但ΔEe比苯的大十余倍;对于T型沥青质二聚体,近距离作用的原子数越多,其ΔEe越大,但近距离作用的原子数只有一个或几个,导致ΔEe明显小于S型;对于PD型沥青质二聚体,RV在约0.35 nm时ΔEe最大(接近S型的2倍),此时二聚体的片层间距明显小于S型,结构最稳定。
参考文献:
[1]MART?N-MART?NEZ F J, FINI E H, BUEHLER M J. Molecular asphaltene models based on Clar sextet theory[J]. RSC Advances, 2015,5(1):753-759.
[2]DOLOMATOV M Y, SHUTKOVA S A, BAKHTIZIN R Z, et al. Structure of asphaltene molecules and nanoclusters based on them[J]. Petroleum Chemistry, 2020,60(1):16-21.
[3]徐春明,刘洋,赵锁奇,等.石油沥青质中杂原子化合物的高分辨质谱分析[J].中国石油大学学报(自然科学版),2013,37(5):190-195.
XU Chunming, LIU Yang, ZHAO Suoqi, et al. Compositional analysis of petroleum asphaltenes by negative ion electrospray high resolution FT-ICR mass spectrometry[J]. Journal of China University of Petroleum (Edition of Natural Science), 2013,37(5):190-195.
[4]WANG H, XU H, JIA W, et al. Revealing the intermolecular interactions of asphaltene dimers by quantum chemical calculations[J]. Energy Fuels, 2017,31(3):2488-2495.
[5]劉必心,龙军,任强,等.塔河沥青质超分子体系的初步探索[J].石油学报(石油加工),2017,33(1):16-24.
LIU Bixin, LONG Jun, REN Qiang, et al.Preliminary exploration for supramolecular system of tahe asphaltene[J]. Acta Petrolei Sinica (Petroleum Processing Section), 2017,33(1):16-24.
[6]任强,龙军,代振宇,等.沥青质分子聚集体中π-π相互作用的研究[J].石油学报(石油加工),2019,35(4):751-758.
REN Qiang, LONG Jun, DAI Zhenyu, et al.Theoretical study on π-π interactions in asphaltene molecular aggregates[J]. Acta Petrolei Sinica (Petroleum Processing Section), 2019,35(4):751-758.
[7]蔡新恒,龙军,任强,等.沥青质分子聚集体的聚集内因[J].石油学报(石油加工),2019,35(5):920-928.
CAI Xinheng, LONG Jun, REN Qiang, et al. Aggregation mechanism of asphaltene molecular aggregates[J]. Acta Petrolei Sinica (Petroleum Processing Section), 2019,35(5):920-928.
[8]PACHECOSANCHEZ J H, ALEJO R, CRUZREYES H, et al. Neural networks to fit potential energy curves from asphaltene-asphaltene interaction data[J]. Fuel, 2019,236(15):1117-1127.
[9]罗辉,邓文安,李传,等.中低温煤焦油沥青质聚集体的分子间作用力[J].中国石油大学学报(自然科学版),2022,46(3):180-187.
LUO Hui, DENG Wenan, LI Chuan, et al. Intermolecular forces of medium/low temperature coal tar asphaltene aggregates[J]. Journal of China University of Petroleum (Edition of Natural Science), 2022,46(3):180-187.
[10]MOUSAVI M, FINI E H. Non-covalent π-stacking interactions between asphaltene and porphyrin in bitumen[J]. Journal of Chemical Information and Modeling, 2020,60(10):4856-4866.
[11]DA COSTA L M, STOYANOV S R, GUSAROV S, et al. Density functional theory investigation of the contributions of π-π stacking and hydrogen-bonding interactions to the aggregation of model asphaltene compounds[J]. Energy fuels, 2012,26(5):2727-2735.
[12]CASTELLANO O, GIMON R, SOSCUN H. Theoretical study of the σ-π and π-π interactions in heteroaromatic monocyclic molecular complexes of benzene, pyridine, and thiophene dimers: implications on the resin-asphaltene stability in crude oil[J]. Energy Fuels, 2011,25(6):2526-2541.
[13]CHAHKANDI M, ALIABAD H A R. Crystalline network form of Gefitinib molecule stabilized by non-covalent interactions: DFT-D calculations[J]. Chemical Physics, 2019,525:110418.
[14]CUTINI M, MASCHIO L, UGLIENGO P. Exfoliation energy of layered materials by DFT-D: beware of dispersion[J]. Journal of Chemical Theory and Computation, 2020,16(8):5244-5252.
[15]DELLEY B. From molecules to solids with the DMol3approach[J]. The Journal of Chemical Physics, 2000,113(18):7756-7764.
[16]DELLEY B. DMol3DFT studies: from molecules and molecular environments to surfaces and solids[J]. Computational Materials Science, 2000,17(2/3/4):122-126.
[17]M?LLER-DETHLEFS K, HOBZA P. Noncovalent interactions: a challenge for experiment and theory[J]. Chemical Reviews, 2000,100(1):143-168.
[18]PANIGRAHI S, BHATTACHARYA A, BANERJEE S, et al. Interaction of nucleobases with wrinkled graphene surface: dispersion corrected DFT and AFM studies[J]. The Journal of Physical Chemistry C, 2012,116(7):4374-4379.
[19]SHERRILL C D, TAKATANI T, HOHENSTEIN E G. An assessment of theoretical methods for nonbonded interactions: comparison to complete basis set limit coupled-cluster potential energy curves for the benzene dimer, the methane dimer, benzene-methane, and benzene-H2S[J]. The Journal of Physical Chemistry A, 2009,113(38):10146-10159.
[20]GRIMME S. Do special noncovalent π-π stacking interactions really exist?[J]. Angewandte Chemie International Edition, 2008,47(18):3430-3434.
[21]GENG Y, TAKATANI T, HOHENSTEIN E G, et al. Accurately characterizing the π-π interaction energies of indole-benzene complexes[J]. The Journal of Physical Chemistry A, 2010,114(10):3576-3582.
[22]LAW K, SCHAUER M, BERNSTEIN E R. Dimers of aromatic molecules: (benzene)2, (toluene)2, and benzene-toluene[J]. The Journal of Chemical Physics, 1984,81(11):4871-4882.
(編辑 刘为清)
