
基于理性设计提高1,3-丙二醇氧化还原酶的稳定性和活性
2023-09-01 00:55付凯璇王雪颖黄彦喆薛海曌胡英菡赵宗保
食品与发酵工业 2023年16期
付凯璇,王雪颖,黄彦喆,薛海曌,胡英菡,赵宗保
1(中国科学院大连化学物理研究所,辽宁 大连,116000)2(中国科学院大学,北京,100000)
1,3-丙二醇(1,3-propanediol,1,3-PD)是重要化工原料,广泛应用于食品、化妆品和医药等领域[1]。根据GB 2760—2014《食品安全国家标准 食品添加剂使用标准》,丙二醇的功能为稳定剂和凝固剂、抗结剂、消泡剂、乳化剂、水分保持剂、增稠剂。目前1,3-PD生产方法主要分为化学法和生物法,与化学合成法相比,生物合成法有着设备成本低、条件温和、环境友好的优点[2]。因此,生物转化,尤其是以廉价原料甘油转化生产1,3-PD,引起广泛关注。目前已在克雷伯氏菌属(Klebsiella)[3]、柠檬酸杆菌属(Citrobacter)[4]、乳杆菌属(Lactobacillus)[5]中发现了将甘油转化为1,3-PD的天然途径。该天然途径涉及两种关键酶,一是甘油脱水酶(glycerol dehydratase, GDHt, EC 4.2.1.30),可将甘油转化为3-羟基丙醛(3-hydroxypropionaldehyde, 3-HPA);二是1,3-丙二醇氧化还原酶(1,3-propanediol oxidoreductase, PDOR, EC 1.1.1.202),在NADH的参与下,将3-HPA还原为1,3-PD(图1-a、图1-b)[6]。来源于肺炎克雷伯菌(Klebsiellapneumoniae)的PDOR(KpPDOR)由于其较高的催化活性和稳定性被广泛研究(图1-c)。KpPDOR适宜pH范围窄,其最适pH值为7.4,而在pH 5.0和pH 10.0时还原活性仅剩40%和20%。温度也是影响KpPDOR活性的因素,在37 ℃还原活性最佳,在20 ℃和55 ℃活性均降低10%[7]。PDOR作为生物合成1,3-PD的限速酶之一,其催化活性,耐温、耐酸碱及稳定性是制约1,3-PD生物制造的重要因素。

a-甘油转化为1,3-丙二醇;b-微生物甘油转化合成1,3-丙二醇途径;c-K.pneumoniae来源PDOR晶体结构
蛋白质定向进化已被广泛应用于提高工业酶在高温、酸碱环境和有机溶剂中的活性和稳定性[8],主要方法包括随机突变、半理性设计和理性设计。随机突变方法需构建大量突变体文库和筛选,JIANG等[9]通过构建K.pneumoniae来源的PDOR的随机突变文库,筛选到氧化活性比野生型PDOR提高4.9倍的A199S突变体。然而,上述方法过程复杂且工作量大。半理性设计方法基于蛋白质结构和序列信息,结合计算机辅助,筛选热点残基,有效提高文库构建效率。PARVEZ等[10]利用序列比对和蛋白结构信息,改造来源于丁酸梭菌(Clostridiumbutyricum) PDOR的二聚体界面氨基酸,经定点饱和突变和筛选,获得突变体P299E,在pH 4.0比野生型PDOR还原活性提高5倍,以及N298C,在pH 8.0下宽温度范围内还原活性提高,但该研究属半理性方法,仍需构建单位点饱和突变文库并筛选上千突变体。理性设计方法通过计算机技术分析蛋白结构和序列信息,指导小而精的定点突变文库设计,提高定向进化效率。目前,生物信息学工具PoPMuSiC 2.1,通过计算蛋白突变的去折叠自由能ΔΔG,预测蛋白质突变后稳定性,其计算相关系数高达0.8[11],已成功辅助改良阿魏酸酯酶[12]、甘油脱水酶[13]和角蛋白酶[14]等蛋白的活性和稳定性。HotSpot Wizard 3.0被广泛应用于识别蛋白的热点氨基酸,以提高蛋白质的稳定性、催化活性、底物特异性等[15],其内置的CAVE、JSD和Fpocket组件可识别位于活性中心且突变后可提高催化功能的可变残基。
本工作通过计算机辅助的方法,结合序列保守性分析,确立KpPDOR定点突变,最终获得催化性能、热稳定性、pH耐受性提高的突变体N262E和V155N。此外,进一步通过蛋白结构模拟探究了KpPDOR性能改良的结构基础。静息细胞转化甘油合成1,3-PD,发现突变体N262E和V155N工程菌株1,3-PD产能高于野生型KpPDOR工程菌株。
1 材料与方法
1.1 材料
1.1.1 菌株和培养基
本研究使用大肠杆菌(Escherichiacoli) BL21(DE3)作为蛋白质表达的宿主,Synbio Technologies股份有限公司合成肺炎克雷伯菌(Klebsiellapneumoniae)来源的PDOR的编码基因dhaT,克隆在质粒pTrc99K-dhaT。通用生物股份有限公司合成丁酸梭菌(Clostridiumbutyricum)来源的GDHt的编码基因dhaB12,克隆在质粒pET15b-dhaB12。2.6节中的工程菌株含有以上2个质粒。
LB培养基:胰蛋白胨10 g/L,酵母提取物5 g/L,氯化钠10 g/L。
TB培养基:胰蛋白胨12 g/L,酵母提取物24 g/L,磷酸氢二钾0.072 mol/L,磷酸二氢钾0.017 mol/L,甘油4 mL/L。
1.1.2 引物
引物由上海生工生物股份有限公司合成,如表1所示。

表1 本研究所用引物
1.1.3 主要试剂和仪器
NADH、3-HPA、甘油,北京鼎国昌盛生物技术公司。
Power Wave XS全波长酶标扫描仪,Bio-Tek Instruments Inc;Molecular Operating Environment(CCG),Montreal;AVANCE Ⅲ核磁共振谱仪(400 MHz),Bruker。
1.2 实验方法
1.2.1K.pneumoniae来源PDOR的理性设计
PoPMuSiC 2.1(http://babylone.ulb.ac.be/popmusic)上传KpPDOR(PDB ID:3BFJ)晶体结构,计算各位点突变后的去折叠自由能(ΔΔG),选取ΔΔG负值显著的作为定点突变。用HotSpot Wizard 3.0 (https://loschmidt.chemi.muni.cz/)内置的CAVE、JSD和Fpocket组件预测功能热点氨基酸,选取可变性分数(mutability score)≥6的氨基酸,并结合BLAST组件分析这些氨基酸的保守性,最终确定突变位点。
1.2.2 突变体的构建和酶活性测定
以质粒pTrc99K-dhaT为模板,通过不依赖限制性内切酶的克隆(RF克隆)方法[16]引入定点突变,PCR产物使用DpnI消化,电转化至E.coliBL21(DE3)感受态细胞中,复苏后,涂布于含卡那霉素的LB平板,37 ℃培养16 h,转化子送上海生物工程有限公司测序。
将构建成功的突变体进行活性鉴定,24孔板培养野生型KpPDOR及其突变体的菌株,每孔2.5 mL LB,50 μg/mL卡那霉素,0.1 mmol/L IPTG,25 ℃诱导48 h,设置野生型KpPDOR为正对照,含pTrc99K空载体的E.coliBL21(DE3)为负对照,设置3个平行;培养结束收集菌体,每孔加入细胞裂解液250 μL[Tris-HCl缓冲液(pH 8.0)10 mmol/L,MgCl21 mmol/L,溶菌酶1 mg/mL,DNaseI 0.1 mg/mL],37 ℃裂解2 h,离心留上清液。酶活性检测体系:37 ℃,N-2-羟乙基哌嗪-N′-2-乙磺酸[4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, HEPES](pH 7.5)50 mmol/L,3-HPA 30 mmol/L,(NH4)2SO430 mmol/L,NADH 0.8 mmol/L,粗酶液10 μL。根据反应液在340 nm光吸收变化测定突变体的活性。
1.2.3KpPDOR和N262E、V155 N的纯化
纯化活性高于野生型KpPDOR的突变体蛋白,将野生型KpPDOR其突变体的菌体使用TB培养,50 μg/mL卡那霉素,0.1 mmol/L IPTG,25 ℃诱导48 h。菌液离心,收集菌体,洗涤后。置于冰浴中超声破碎,离心收集裂解液上清,上清液倒入Ni-NTA层析柱,先后以洗涤缓冲液NWB(50 mmol/L NaH2PO4,0.5 mol/L NaCl,40 mmol/L咪唑,pH 8.0)洗涤杂蛋白,以洗脱缓冲液NEB(50 mmol/L NaH2PO4,0.5 mol/L NaCl,250 mmol/L咪唑,pH 8.0)洗脱,收集蛋白溶液,于超滤离心管中超滤浓缩。用Bradford方法以BSA为标准品测定蛋白质浓度。在12%变性SDS-PAGE凝胶中分析纯化的N262E和V155N蛋白,以BeyoColorTM彩色预染蛋白分子质量标准(10~170 kDa)确定目的蛋白分子质量。
1.2.4 酶活力的测定
还原反应酶活性检测体系37 ℃,HEPES(pH 7.5) 50 mmol/L,3-HPA 30 mmol/L,(NH4)2SO430 mmol/L,NADH 0.8 mmol/L,野生型和突变体纯酶0.56 μg/mL。每个样品设置3个平行。氧化反应测定体系为37 ℃,HEPES(pH 7.5) 50 mmol/L,(NH4)2SO430 mmol/L、NAD 2 mmol/L,酶0.52 μg/mL,1,3-PD 100 mmol/L。根据反应液在340 nm光吸收变化测定突变体的活性。酶活力单位的定义:每分钟催化产生1 μmol NADH所需的酶量。NADH的摩尔消光系数为6.22 L/(mmol·cm)。
1.2.5 酶学性质研究
测定突变体在pH 2.0~12.0的酶活力,所用缓冲液为甘氨酸-盐酸缓冲液(pH 2.4) 100 mmol/L、乙酸-乙酸钠缓冲液(pH 4.0) 100 mmol/L、HEPES缓冲液(pH 7.5) 50 mmol/L、甘氨酸-氢氧化钠缓冲液(pH 10.0) 50 mmol/L和氯化钾-氢氧化钠缓冲液(pH 12.0)200 mmol/L。检测体系3-HPA 30 mmol/L、(NH4)2SO430 mmol/L、NADH 0.8 mmol/L、酶0.56 μg/mL,酶在37 ℃分别与pH 2.0~12.0的缓冲液孵育2 min后,加入3-HPA和NADH启动反应。根据反应液在340 nm光吸收变化测定突变体的活性。每个样品设置3个平行。研究酶的pH稳定性。酶在37 ℃分别与pH 2.0~12.0的缓冲液孵育1 h后,测定酶活力,以孵育2 min酶活力为100%,每个样品设置3个平行。
测定突变体在温度25~55 ℃的酶活力,检测体系HEPES(pH 7.5) 50 mmol/L,3-HPA 30 mmol/L、(NH4)2SO430 mmol/L、NADH 0.8 mmol/L和酶0.56 μg/mL。酶在pH 7.5,25~55 ℃孵育2 min后,加入3-HPA和NADH启动反应。根据反应液在340 nm光吸收变化测定突变体的活性。每个样品设置3个平行。研究酶的温度稳定性,酶分别在pH 7.5,25~55 ℃孵育1 h后,测定酶的残余活力,以孵育2 min 酶活力为100%。每个样品设置3个平行。
将纯化后的KpPDOR及N262E,V155N在pH 7.5,37 ℃孵育,于0、30、60、120、180、240、300、330 min取样并测定残余酶活力。以孵育2 min酶活力为100%。将残余酶活力的 ln值为对时间t作图,计算酶失活速率常数k,由公式t1/2=ln2/k,计算酶的半衰期。
1.2.6 酶动力学
还原反应测定体系为HEPES(pH 7.5) 50 mmol/L,(NH4)2SO430 mmol/L、NADH 0.8 mmol/L,酶0.52 μg/mL,3-HPA梯度浓度,加超纯水至100 μL,每个样品设置3个平行。将酶在37 ℃,pH 7.5孵育2 min后。加入3-HPA和NADH启动反应。根据反应液在340 nm光吸收变化测定突变体的活性。氧化反应测定体系为HEPES (pH 7.5) 50 mmol/L,(NH4)2SO430 mmol/L、NAD 2 mmol/L,酶0.52 μg/mL,1,3-PD梯度浓度,方法同上,每个样品设置3个平行。使用Lineweaver-Burk双倒数作图法进行数据处理,计算动力学常数。
1.2.7 酶结构分析
使用分子操作环境(molecular operating environment,MOE)分别进行KpPDOR野生型和突变体的结构分析。选择KpPDOR的晶体结构的A链作为模板,在A链的基础上进行定点突变,并能量最小化。
1.2.8 静息细胞催化
1.2.8.1 样品准备
将1.1.1节中的质粒pET15b-dhaB12通过电转化的方法分别转入含有KpPDOR野生型和突变体质粒的E.coliBL21(DE3)中,构建工程菌株。将KpPDOR野生型和突变体的工程菌接入LB培养基(50 μg/mL卡那霉素和100 μg/mL羧苄青霉素)接菌,37 ℃培养12 h,以初始OD600值为0.05转接于50 mL LB培养基(50 μg/mL卡那霉素,50 μg/mL羧苄青霉素 0.2 mmol/L IPTG),25 ℃诱导48 h。取7.5×109个细胞,用50 mmol/L HEPES缓冲液(pH 7.5)洗1次,然后用0.5 mL静息细胞反应液(50 mmol/L HEPES,40 mmol/L 甘油)重悬,37 ℃、200 r/min反应2 h。加入等体积0.1 mol/L HCl终止,离心,取上清液。将样品冷冻干燥,后溶于0.5 mL氘水中,加入顺丁烯二酸使其终浓度为60 mmol/L。
1.2.8.2 定量核磁检测产物
采用定量核磁氢谱方法(quantitative nuclear magnetic hydrogen spectroscopy,1H-qNMR)检测1,3-PD含量,为了精准定量,选取合适的内标顺丁烯二酸,其在氘水溶剂中化学位移6.3且不与样品峰重合。设置D1为30 s以保证目的氢核驰豫充分;扫描次数NS为32以保证信噪比。
1.2.8.3 数据处理
将图谱先进行基线和峰形的校正。根据文献[17],1.7~1.8的五重峰归属与1,3-PD C2的2个氢原子,因此使用1.7~1.8处峰定量1,3-PD,利用核磁氢谱不同化学位移的氢峰面积与氢核数目成正比,用已知含量的顺丁烯二酸作为定量标准,对1,3-PD进行定量。
2 结果与分析
2.1 理性设计定向突变
PoPMuSiC 2.1预测的结果(图2)显示了每个残基的最优突变及其对应的去折叠自由能(ΔΔG),从中选择ΔΔG负值显著的L38E、V99H、G144T、V155N作为定点突变位点(图2-c)。HotSpot Wizard 3.0内置的CAVE、JSD和Fpocket组件预测功能热点氨基酸,选取可变性分数≥6的氨基酸作为候选位点(图2-a),结合内置的BLAST组件分析候选位点序列保守性(图2-b),其第276、272、283和262位氨基酸出现频率最高的分别是Phe、Pro、Leu、Gly和Glu。综上,构建L38E、V99H、G144T、V155N、N262E、N262G、L276F,Q272P,V283L共9个突变体(表2)。

表2 KpPDOR定点突变位点

a-HotSpot Wizard 3.0功能热点氨基酸分析;b-HotSpot Wizard 3.0序列保守性分析;c-PoPMuSiC 2.1去折叠自由能分析;d-最终选定的突变位点
2.2 突变体的构建和活性鉴定
由RF克隆成功构建点突变后的质粒(图3-a),转入E.coliBL21(DE3)中。测定KpPDOR及突变体在37 ℃,pH 7.5的对3-HPA的还原活性,结果如图3-b所示,N262E的还原活性是KpPDOR的1.1倍,V155N是KpPDOR的1.7倍。其余突变体活性均低于KpPDOR。将野生型KpPDOR、N262E、V155N通过Ni-NTA蛋白纯化系统纯化,并进行SDS-PAGE鉴定,KpPDOR、N262E、V155N纯化后蛋白无杂带,分子质量约为42 kDa(图3-c)。

M-蛋白分子量标准;泳道1-WT;泳道2-N262E;泳道3-V155N
2.3 酶学性质探究
2.3.1 酶在不同pH的活性和稳定性
分别检测了KpPDOR及其突变体在pH 2.0~12.0条件下的还原活性(图4-a),KpPDOR最适pH 7.5,N262E、V155N的最适pH与KpPDOR一致。酸性条件(pH 4.0)下,V155N和N262E的酶活性分别为KpPDOR的2.5倍和1.9倍。碱性条件(pH 10.0)下,V155N酶活性为KpPDOR的1.6倍。因此,V155N在pH 4.0~10.0,N262E在pH 4.0~7.5活性高于KpPDOR,这表明V155N和N262E能适应更宽泛的pH条件。

a-KpPDOR和突变体在25~55 ℃的活性;b-KpPDOR和突变体在25~55 ℃的热稳定性;c-KpPDOR和突变体在pH 2.0~12.0时的活性;d-KpPDOR和突变体在pH 2.0~12.0时的稳定性;e-KpPDOR和突变体在37 ℃时的半衰期;f-KpPDOR和突变体的Michaelis-Menten方程
pH稳定性的结果显示(图4-b),在pH 2.0~12.0孵育1 h后,KpPDOR分别保留57%、72%、76%、73%、64%、59%的残余酶活力,V155N和N262E在相同条件下处理后,V155N和N262E残余酶活力略高于KpPDOR,pH稳定性优于KpPDOR。
2.3.2 酶在不同温度的活性和稳定性
分别检测了KpPDOR及其突变体在25~55 ℃条件下的活性(图4-c),KpPDOR最适温度为37 ℃,N262E、V155N与KpPDOR一致。V155N在25、37、45、55 ℃酶活性分别是是KpPDOR的1.3、1.8、1.2、1.4倍。这表明V155N在不同温度下保持更高的催化活性。
酶热稳定性结果显示(图4-d),在25~55 ℃孵育1 h后,N262E分别保留87%、90%、84%、74%的残余酶活力,高于KpPDOR,V155N在25~55 ℃残余酶活力略高于KpPDOR。因此,N262E和V155N热稳定性优于KpPDOR。
2.3.3 酶半衰期测定
测定KpPDOR和V155N、N262E在37 ℃,pH 7.5孵育0~300 min的残余酶活力(图4-d)。结果表明,在孵育120 min后,KpPDOR保留71%的酶活力,V155N和N262E分别保留78%和84%的残余酶活力;在孵育240 min后,野生型KpPDOR保留56%的残余酶活力,而V155N和N262E分别保留酶活力的64%和71%。N262E在37 ℃的半衰期为462 min,为KpPDOR的1.4倍,V155N的半衰期为365 min,为KpPDOR的1.1倍(图4-e)。
2.4 酶动力学
测定KpPDOR和V155N、N262E的对还原反应的底物3-HPA和氧化反应底物1,3-PD的动力学数据(表3)。KpPDOR比酶活51.16 U/mg,V155N和N262E比酶活分别为72.08 U/mg和60.22 U/mg,分别比KpPDOR提高41%和18%。从还原反应来看,V155N和N262E的Km分别为KpPDOR的0.59倍和0.85倍,表明其对3-HPA结合亲和力提高,催化效率kcat/Km分别为KpPDOR的2.7倍和1.49倍。氧化反应与还原反应的数据结果相互印证。

表3 野生型和突变体动力学
2.5 酶的结构分析
MOE分析野生型KpPDOR和V155N、N262E蛋白结构,如图5所示。N取代155位上的V后,蛋白结构引入两个氢键,V155N侧链氮原子与D106侧链羧基的氧原子形成距离2.01 Å的氢键;侧链氧原子与K164侧链氮原子形成距离2.72 Å的氢键(图5-b)。氢键作为一种强非共价相互作用力,可稳定155位氨基酸所在的β-折叠,从而提高酶稳定性[18]。E取代262位N,N262E的羧基与R152上胍基的两个氮原子形成距离分别为3.72 Å和3.97 Å的氢键(图5-d),增强了催化中心的刚性[19]。

a-野生型V155位点与周围氨基酸相互作用;b-突变体V155N位点与周围氨基酸相互作用;c-野生型N262位点与周围氨基酸相互作用;d-突变体N262E位点与周围氨基酸相互作用
2.6 静息细胞转化结果
通过静息细胞转化,以甘油为底物转化合成1,3-PD,以1H-qNMR方法对野生型KpPDOR和突变体V155N、N262E的工程菌1,3-PD产量进行定量,该方法具有快速便捷的优点[20]。图6为不同菌株反应后产物的核磁共振氢谱,用已知含量的顺丁烯二酸作为定量标准,以1.7~1.8的五重峰[17]对1,3-PD进行定量。

a-野生型KpPDOR反应后的谱图;b-N262E反应后的谱图;c-V155N反应后的谱图;d-BL21(DE3)负对照反应后的谱图
经数据处理,各体系中1,3-PD含量如表4所示。将顺丁烯二酸的峰面积归一化为2。经2 h的静息细胞转化后,野生型KpPDOR转化后1,3-PD含量为12.3 mmol/L,产能0.82 mmol/L/OD600。突变体N262E和V155N转化后的1,3-PD产能分别提升至0.90 mmol/L/OD600和1.16 mmol/L/OD600。这说明突变体对甘油生物转化为1,3-PD具有促进作用。
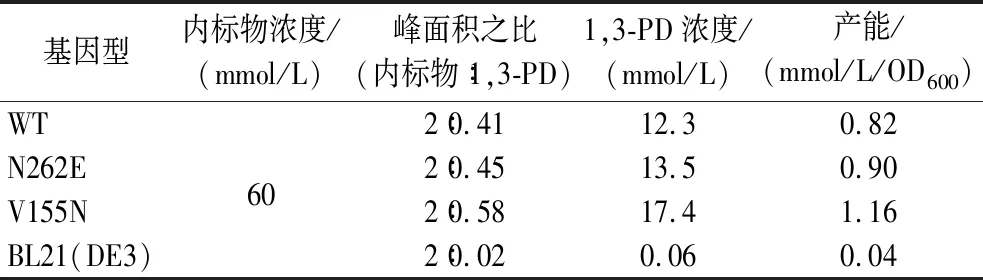
表4 1H-qNMR数据处理结果
3 结论与讨论
1,3-PD在食品领域常作为加工助剂、吸湿剂和溶剂。在备受关注的甘油生物转化合成1,3-PD中,KpPDOR是其中的关键限速酶之一,因此,其催化活性、热稳定性和pH耐受性的改良有重要价值。本研究改造KpPDOR的策略,一方面借助PoPMuSiC 2.1计算蛋白质热稳定性的重要指标去折叠自由能ΔΔG,一方面借助Hotspot wizard 3.0功能热点分析和保守序列分析,理性选取定点突变位点。最终成功获得性能改良的N262E和V155N突变体,其可促进甘油转化为1,3-PD。这说明其具有应用于1,3-PD生物制造的潜力。
本工作获得25~55 ℃酶活力高于KpPDOR的V155N,以及热稳定性高于KpPDOR的N262E,这拓宽了该酶在工业生产中宽温度范围的活性和稳定性。此外,宽pH范围的活性和稳定性对1,3-PD合成也是至关重要的。有研究表明,由于微生物发酵产1,3-PD 积累有机酸,导致胞内pH值的下降会抑制KpPDOR活性,降低1,3-PD产量[21]。此外,有研究报道,pH 6.5时体外合成1,3-PD的产率是中性条件下的2.2倍[22]。这表明弱酸性环境有利于体外合成1,3-PD合成,因此提高KpPDOR耐酸性具有必要性。V155N pH 4.0的酸性条件下催化活性为KpPDOR的2.5倍,且同时在更宽温度范围活性高于KpPDOR。综上分析表明,V155N具有应用于微生物内和体外环境生产1,3-PD的潜力。
本工作通过蛋白结构分析,加深了对蛋白结构和功能的认识。氢键作为蛋白最重要的非共价相互作用力,对维持蛋白质的二级、三级结构起到关键作用[23]。V155位于KpPDOR的β-折叠,N取代155位上的V,引入两个氢键,与其形成氢键的D106和K164分别位于KpPDOR的α-螺旋和β-折叠(图4-b),氢键稳定了该区域的二级结构,从而使V155N在较宽的pH和温度范围内的维持较稳定的构象,以发挥其活性。有研究表明,改造酶活性中心结构灵活的残基可提高酶稳定性和活性,如引入氢键可提高催化中心刚性,使蛋白更稳定[19]。KpPDOR催化中心的262位点引入两个氢键(图4-d),提高其刚性,利于其在极端条件下保持催化中心的正确构象[24]。体外实验结果与以上结构分析表现出一致性。如需进一步阐明KpPDOR构效关系,可结合X射线衍射、核磁共振和远、近紫外光谱等手段来研究蛋白结构。
综上所述,基于理性设计的方法指导KpPDOR改造,成功改良其催化活性、热稳定性和耐酸性,有望提升1,3-PD的生物制造的产能,从而降低其在食品工业中的成本。获得性能优化的突变体后,进一步通过结构分析阐释了性能优化的结构基础,加深了对生物酶构效关系的认识。本研究使用的理性设计的思路,也为改良其他工业酶的性能提供了指导。
猜你喜欢
中国科技纵横(2021年24期)2021-03-02
中成药(2018年6期)2018-07-11
能源(2017年7期)2018-01-19
中学科技(2017年11期)2017-12-26
安徽医科大学学报(2016年12期)2017-01-15
山东农业工程学院学报(2016年6期)2016-12-01
中国民族医药杂志(2016年4期)2016-05-09
天津医科大学学报(2015年2期)2015-12-22
山东医药(2015年40期)2015-02-28
化工生产与技术(2014年4期)2014-02-27
