
氟硼吡咯类(BODIPY)三重态光敏剂在光催化中的应用
2023-08-24 05:07:28王艳红刘仲能
工业催化 2023年8期
董 羽,刘 旭,王艳红,余 强,刘仲能
(中石化(上海)石油化工研究院有限公司,上海 201208)
随着经济的飞速发展,作为经济社会发展的引擎,世界各国对于能源的需求与日俱增,而化石能源在全世界能源消耗量的占比超过85%[1]。由于对化石能源的过度依赖会产生诸如国家能源安全、环境污染、可持续性等一系列问题,世界各主要经济体均大力发展可替代化石能源的新能源。太阳能作为人类取之不尽用之不竭的清洁能源,是人类解决能源危机的重要途径。因此,如何对太阳能进行有效利用,高效转化为人类社会能够直接使用的电能、化学能等形式的能量,是今后科技发展的重要方向。
近年来,将光作为能量,以对光敏感的物质作为催化剂,对有机底物进行催化合成的反应受到科技工作者的关注[2-5]。光催化领域的发展正日益加快,涉及许多不同的科学研究领域,在诸如可再生能源和化学原料、新反应开发、天然产物合成、材料和生物应用等领域扮演着重要的角色[4,6-8]。
光催化反应的核心在于催化体系的设计,而催化体系的核心在于催化剂的设计与合成。光催化反应所使用的催化剂均为对光敏感的物质,此类化合物受光激发后,可以高效产生高能量的激发态,并能够将激发态的性质(如电子、能量或自旋等)传递给反应体系中的反应物或者氧分子等反应底物,因此也被称为光敏剂。三重态光敏剂是光敏剂中很重要的一类化合物,这类分子受光激发后,可以通过系间窜越(ISC)产生电子自旋多重度为3的激发态,即三重激发态,并能够将三重激发态的性质传递给其他不能够自发产生三重态的分子。三重态光敏剂在光解水产氢[9-11]、光催化有机合成反应[12-13]、光致还原CO2[14-15]等领域有着广泛的应用。三重态光敏剂在反应体系中通常通过与底物发生单电子电子转移或能量转移实现催化性能[16]。
理想的三重态光敏剂应当具有以下的性质:(1)在可见光区具有较强的吸收,即摩尔消光系数要大,有利于对可见光区能量的高效利用;(2)具有好的光稳定性,以保证在光照催化过程中,光敏剂不出现光漂白现象;(3)具有高的三重态量子效率,可以有效获得具有更好催化效能的三重激发态;(4)基于电子转移催化机理的光敏剂要具有可逆的氧化还原电位,以保证光敏剂在催化循环过程中的稳定性和可重复性。基于能量转移催化机理的光敏剂要具有较高的三重态能级,以利于能量转移过程的发生;(5)光敏剂的三重激发态要具有较长的三重态寿命,以保证光敏剂分子有足够的时间可以将自身的三重态性质通过分子间的能量转移或电子转移过程传递给底物;(6)光敏剂母体发色团本身应当具有良好的可衍生化能力,有利于根据不同类型的反应进行光敏剂的模块化设计。
目前,贵金属配合物是在光催化领域应用最广泛的三重态光敏剂,如Ru(Ⅱ)[17-18]、Ir(Ⅱ)[19-20]、Au(Ⅰ)[21-22]等,如图1所示。这一类光敏剂的优势在于,基于贵金属原子的重原子效应,可以高效产生三重激发态用于光催化,而且此类配合物激发态的氧化还原电位适用于电子转移机理的光催化反应。但是这一类配合物仍然存在一些不足:(1)对可见光的吸收起主要作用的一般是基于金属到配体的吸收(MLCT),这种吸收的摩尔消光系数一般较小,不利于对光子的高效利用,如三联吡啶钌1-1在450 nm处的摩尔消光系数约为15 000 M-1·cm-1[23],而三(2-苯基吡啶)铱1-3在400 nm处约为5 000[24];(2)Ru、Ir、Au等重金属价格昂贵,不利于控制催化剂的成本;(3)贵金属原子的重原子效应很强,不仅会促进单重态跃迁到三重态的ISC,也会促进三重态跃迁回基态的ISC过程,因此三重态的寿命一般较短,不利于扩散控制分子间的能量转移或电子转移,如化合物1-1的三重态寿命仅为0.87 μs[25]。因此,纯有机三重态光敏剂的研究价值便凸显出来。这一类光敏剂的优势在于:不含有贵金属,且价格优势明显;摩尔消光系数大,对可见光的利用效率更高;可实现高效ISC的方式多(如重原子效应、自由基诱导、电荷分离再复合等方式),可以根据应用场景进行选择;发色团种类多且衍生化能力强,有利于根据不同的反应需要进行模块化设计等。
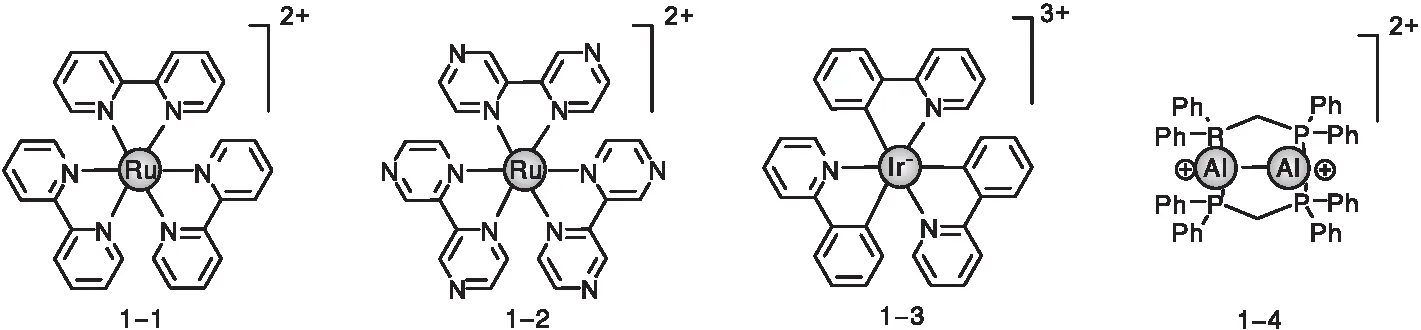
图1 含有贵金属的光催化剂Figure 1 Photocatalysts containing precious metal
1 光催化反应的光化学理论基础
1.1 光敏剂的光物理过程
光敏剂受光激发后的光物理过程可通过Jablonski能级图进行描述,如图2所示。光敏剂分子受光激发后,通过垂直跃迁到达高能级的单重激发态(Sn,n≥2)或者第一单重激发态的高振动能级,随后分子会通过内转换(IC)或者振动弛豫的方式回到第一单重激发态(S1),单重态分子可以通过荧光发射或者非辐射跃迁的方式回到基态。尽管ISC过程为自旋禁阻,通常不会主动发生,但是三重态光敏剂可以通过ISC过程到达与之能量最接近的三重激发态(Tm,m≥1),同时ISC过程是需要其他条件的促进才能高效发生。

图2 Jablonski能级示意图[26]Figure 2 Simplified diagram of Jablonski energy level[26]
常见的促进ISC方式有:重原子效应、电荷分离再复合、自由基诱导、单重态裂变等。高能级的三重激发态会通过IC过程到达第一三重激发态(T1),T1态会通过磷光发射或者非辐射跃迁的过程回到基态,在此过程中,由于T1→S0的跃迁为自旋禁阻,因此磷光寿命和三重态寿命均远长于相应的单重态寿命。而T1态则是三重态光敏剂实现催化性能最重要的分子态。
1.2 光敏剂应用于催化反应的机理
光敏剂作为光催化剂应用于催化反应中的机理主要有3种:光致电子转移、光致能量转移、光致产生活性氧机理。
光致电子转移和光致能量转移机理如图3所示[16]。

图3 三重态光敏剂催化过程示意图(a)单电子转移机理;(b)能量转移机理[16]Figure 3 Schematic representation of photocatalysts in photocatalytic process (a)single electron transfer mechanism and (b)energy transfer mechanism[16]
1.2.1 光致电子转移
光致电子转移通常为单电子转移机理,该机理分为氧化猝灭和还原猝灭两种途径,如图3(a)所示。氧化淬灭途径为:光敏剂/光催化剂(PC)受光激发后到达激发态(PC*),激发态分子传递出一个电子给电子受体形成光敏剂分子的阳离子自由基(PC•+),电子受体则得到一个电子形成阴离子自由基中间体(A•-),电子受体可以是缺电子的底物或者质子还原催化。光敏剂分子的阳离子自由基与电子牺牲试剂发生第二次单电子转移,将光敏剂分子还原为基态。还原淬灭途径为:光敏剂(PC)受光激发后到达激发态(PC*),富电子的底物或电子牺牲试剂将一个电子传递给光敏剂的激发态分子,形成光敏剂分子的阴离子自由基(PC•-)和一个阳离子自由基中间体(D•+),光敏剂分子的阴离子自由基再与催化剂发生电子转移反应,将光敏剂分子还原为基态。
1.2.2 电子转移的热力学与动力学
单电子转移是否能够发生,可以通过计算该电子转移体系的热力学进行判定,即计算电子转移的吉布斯自由能变(ΔGET)。该值可以通过Rehm-Weller方程进行计算,见式(1)~(2)[27]:
ΔGET=e[EOX-ERED]-E00+ΔGS
(1)
(2)
式中,ΔGS为静态库仑能;e为电子电荷;EOX为电子给体的单电子氧化半波电位;ERED为电子受体的单电子还原半波电位;E00表示分子的激发态能级;ε0=8.85×10-12F·m-1,为真空介电常数;εS为需要计算的溶剂的介电常数;εREF为电化学测试时使用的溶剂的介电常数;RCC是电子给体和受体之间的距离(在均相催化反应中,电子给受体之间为分子间的电子转移,因此RCC的值通常可认为是无穷大);RD和RA分别是电子给体和电子受体的半径。
当计算得到的电子转移吉布斯自由能变ΔGET为负值时,证明该体系发生电子转移是热力学允许的,反之则为热力学禁阻。
在经典体系中,电子转移动力学可以通过Marcus理论进行描述,电子转移速率与电子转移驱动力之间的关系见式(3)[28-29]:
(3)
式中,kET代表电子转移的速率常数,V为发生电子转移的两个态之间的电子耦合矩阵元,λ是重组能,重组能λ由体系内部的重组能(λi)和体系外部介质对体系影响的重组能(λs)两部分组成,kB为玻尔兹曼常数,ΔGET为电子转移的吉布斯自由能变。
由式(3)可以得到电子转移的速率kET与电子转移驱动力ΔGET之间的关系图(图4)。
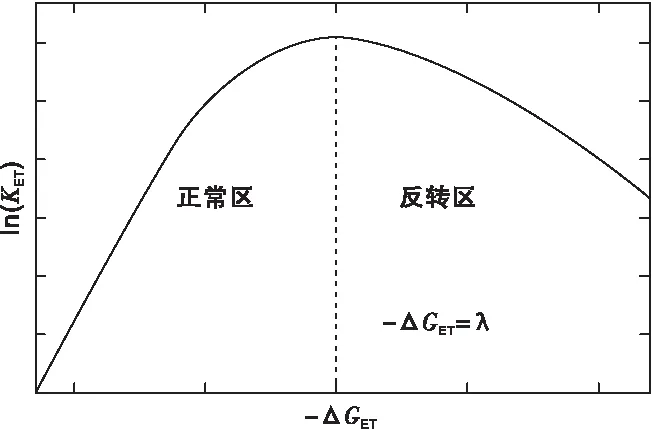
图4 电子转移速率与电子转移吉布斯自由能变的关系Figure 4 Relationship between electron transfer free energy change and electron transfer rate
由图4可知,电子转移速率与电子转移驱动力并非线性关系,而是呈现类似倒抛物线的曲线关系,当-ΔGET小于重组能λ时,kET与-ΔGET呈正比,即驱动力越大,电子转移速率越快,此时处于正常区;当-ΔGET大于重组能λ时,kET与-ΔGET呈反比,即驱动力越大,电子转移速率越慢,此时处于反转区;当-ΔGET等于重组能λ时,电子转移速率最快,达到峰值。但是Marcus反转区仅在电子给受体之间距离较小时、分子内电子转移时才能够存在,而在均相中的分子间电子转移体系不会观察到。原因是当-ΔGET增加到一定值时,电子转移速率会达到扩散控制而保持不变[30]。
1.2.3 光致能量转移
光致能量转移机理如图3(b)所示,光敏剂分子受光激发后到达单重激发态(1PS*),单重激发态通过高效的ISC过程实现三重激发态的有效布居(3PS*),因为三重态的寿命较长,可以与底物分子发生Dexter机制的能量转移,获得底物的激发态(EA*),进而发生相应的化学反应。
1.2.4 光致产生活性氧

图5 光致产生活性氧的机理[36]Figure 5 Schematic representation of photoinduced production of reactive oxygen species[36]
2 氟硼吡咯类(BODIPY)化合物在光催化中的应用
2.1 氟硼吡咯类(BODIPY)化合物简介
氟硼吡咯类(BODIPY)化合物于1968年被科学家Treibs A和Kreuzer F H首次报道合成[40]。该类化合物的母体结构是由吡咯与氟硼络合而成的共轭体系(图6),这种共轭结构刚性很强,使得BODIPY的吸收和发光谱带宽度较窄。更重要的是,这一类结构赋予了BODIPY分子良好的可衍生化能力,BODIPY母体结构上有8个衍生化位点,应用最广泛的是在8位、2位以及6位通过取代反应、偶联反应以及点击化学等方式,依据应用需求衍生化出一系列功能化化合物[41-43]。
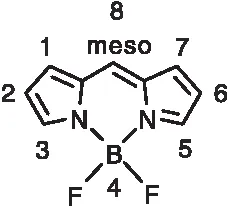
图6 BODIPY的母体结构Figure 6 Structure of BODIPY molecule
顾培洋教授课题组总结了BODIPY研究的历史脉络(图7)[43],由于其良好的荧光性质,首先被应用于荧光探针领域,随后拓展到光动力治疗[36]、上转换[42]、光催化[16]等领域。
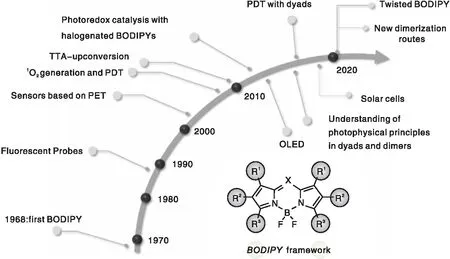
图7 BODIPY研究进展的历史脉络图[43]Figure 7 Historical development of BODIPY in various applications[43]
2.2 氟硼吡咯类(BODIPY)光敏剂在光催化领域的应用
2.2.1 含有过渡金属的BODIPY类光敏剂
过渡金属类配合物光敏剂在可见光区的主要吸收为MLCT吸收,即金属-配体电荷转移吸收,这种吸收的摩尔消光系数较小、三重态寿命较短,限制了其进一步的应用。通过在配体上修饰吸光能力强且三重态寿命长的有机发色团,能将由于轨道重叠程度小导致的S0→1MLCT的弱吸收转变为S0→1π-π*的强吸收,既不会削弱金属原子的重原子效应,又能保证三重激发态的产率,而且可以将短寿命的3MLCT转化为长寿命的3IL(配体三重态)。BODIPY作为既具有强可见光吸收也具有长三重态寿命的发色团,可以作为过渡金属配合物的配体,以期改善光敏剂的光化学性质。
赵建章教授课题组将BODIPY作为配体分别以不共轭(3-1)和共轭(3-2)两种方式连接到Ir配位中心上(图8)[34],并测试了传统的Ir配合物1-3、光敏剂3-1和3-2的单线态氧量子产率(ΦΔ),分别为92%、52%和97%,单线态氧量子产率可以作为衡量光敏剂ISC能力的参数指标;3种配合物的三重态寿命分别为0.35 μs、23.7 μs和87.2 μs,相较于配合物1-3,光敏剂3-2的三重态寿命增加了约250倍,同时通过瞬态吸收光谱和自旋密度计算证明1-3和3-2的三重态分别为3MLCT和3IL。由此可见,将BODIPY以共轭的形式作为配体的一部分,与金属中心直接作用更有助于提升Ir的重原子效应,实现高效ISC的同时,将短寿命的3MLCT转化为长寿命的3IL。

图8 含BODIPY作为配体的Ir配合物的分子结构Figure 8 Structures of Ir-complexes containing BODIPY as ligand
胡桃醌(Juglone)是一种抗菌消炎药物,也是抗肿瘤药物N-苯胺取代萘醌类物质的重要中间体,可以通过光催化氧化1,5-二羟基萘(DHN)的方式获得[44]。光敏剂光催化氧化1,5-二羟基萘制备胡桃醌的机理如图9所示,光敏剂受光激发后产生三重激发态,激发态分子敏化三线态氧生成具有高氧化活性的单线态氧,单线态氧对1,5-二羟基萘进行类似D-A环加成的反应,最后脱水生成胡桃醌[45]。将光敏剂1-3、3-1和3-2应用于该反应中,发现在相同反应条件下,3-2体系的1,5-二羟基萘转化速率约为3-1的4倍、1-3的11倍,经过40 min光照之后,3-2体系胡桃醌产率分别约为3-1的1.3倍、1-3的2.4倍。证明了长寿命、高ISC能力的光敏剂在光催化氧化反应上的优势。

图9 光催化氧化1,5-二羟基萘制胡桃醌的机理(单线态氧)[45]Figure 9 Schematic representation of photocatalytic oxidation of 1,5-dihydroxynathalene (DHN) to juglone (singlet oxygen)[45]
基于上述策略,Wang P等设计并合成了光敏剂3-3(图10)[46],将在可见光区有强吸收的BODIPY和香豆素发色团以配体的形式参与到Ir(Ⅲ)原子的配位中,实现了(400~575) nm的宽谱带吸收,几乎覆盖了可见光区的一半。并且依靠Ir的强重原子效应实现了高效的ISC,获得89 μs的长寿命三重态。基于上述性质,将其应用于光催化产氢实验中:以3-3为光敏剂、CoⅢ(dmgH)2(py)Cl为催化剂、N,N-二甲基对甲苯胺(DMT)为电子牺牲试剂,宽谱带氙灯为光源,实现了高效的光催化产氢。其中光敏剂3-3的TON值可达115 840,是传统Ir配合物1-3的21倍,催化效果为1-3的320倍,充分证明宽谱带吸收长寿命的三重态光敏剂在光催化产氢领域的应用潜力。

图10 含BODIPY作为配体的Re和Pd配合物的分子结构以及所催化的反应通式(a) 1,5-二羟基萘光催化氧化制胡桃醌;(b) Sonogashira偶联反应Figure 10 Structures of Re and Pd-complexes containing BODIPYas ligand and photocatalytic reactions over them (a)photocatalytic oxidation of 1,5-dihydroxynathalene (DHN) to juglone; (b) Sonogashira coupling reaction
Wang D等[47-48]利用BODIPY的结构,将其与金属Ir进行共价连接,获得二元BODIPY-Ir化合物(化合物3-4~3-6,图8)。该系列化合物的ISC能力明显增强(ΦΔ值最高为10.4%),并成功拓展到光催化氢化胺化、烷氧化以及硫醚催化氧化反应中。
除金属Ir之外,上述的分子设计策略也可以应用于金属Re和Pd的配合物设计中(图10)。赵建章教授课题组[49]将金属Re与BODIPY-联吡啶配体络合,得到了光敏剂3-8,与母体Re配合物3-7相比,三重态寿命从26 ns延长为104 μs,并将ΦΔ提升至88%。在光催化氧化1,5-二羟基萘制胡桃醌的反应中,光敏剂3-8体系在光照40 min后的产率为3-7的9倍。Dissanayake K C等[50]合成了以Pd为催化活性中心的化合物3-9,并成功应用于Sonogashira偶联反应中,拓展了光敏剂的催化反应类型。在室温下,以绿光LED为光源,光照24 h后以90%以上的产率得到碘代苯与苯基炔类化合物偶联的产物。但是该催化体系具有底物局限性,例如在碘代苯的对位含有吸电子取代基时,产率会大幅下降(硝基和氰基取代物的产率分别为18%和33%)。同时,作者认为光敏剂3-9激发态发生的能量转移过程有利于催化循环中的还原消除步骤,是室温下能够加快该反应催化进程的主要原因。
由于Ir、Re、Pd等均为稀有金属,价格昂贵且储量稀少,因此需要拓展更多可用于光催化的廉价金属配合物。张志明和郭颂等[51]报道了以Cu作为配位中心、BODIPY为光吸收天线的光敏剂3-11(图11),作者并未将BODIPY直接与Cu原子进行配位,而是通过两次能量传递的方式实现高效的ISC:BODIPY吸收光子到达单重激发态后发生能量转移(TBET),获得Cu配合物的单重激发态,Cu配合物发生ISC获得其三重激发态,通过三重态-三重态能量传递(TTET)过程获得长寿命的BODIPY三重激发态。

图11 含BODIPY作为配体的Cu配合物的分子结构以及所催化的反应通式(a) 1,5-二羟基萘光催化氧化制胡桃醌;(b) 芳基硼酸催化氧化制酚Figure 11 Structures of Cu-complexes containing BODIPYas ligand and photocatalytic reactions over them (a) photocatalytic oxidation of 1,5-dihydroxynathalene (DHN) to juglone;(b)photocatalytic aerobic oxidation of aromatic boronic acids to phenols
该策略充分利用了BODIPY强吸光和长三重态寿命的性质,并且不依赖于直接的发色团-金属配位,即可获得长三重态寿命(3-11的三重态寿命为27.2 μs,为3-10的100倍)。作者将光敏剂3-11分别应用于能量转移机理的1,5-二羟基萘光催化氧化制胡桃醌以及单电子转移机理的芳基硼酸光催化氧化制酚反应中,相同反应条件下,3-11体系光催化氧化1,5-二羟基萘的反应速率分别为1-1和1-3的14.8和15.7倍。3-11体系酚的光催化产率为3-10的4.85倍,而且在底物(如芘硼酸、4-甲基硼酸、4-叔丁基硼酸等)适用性上强于传统光敏剂1-1和1-3。这种分子设计策略可以简化配体的合成并拓展配体的种类。
2.2.2 基于碘、溴重原子效应的BODIPY类光敏剂
虽然稀有金属的重原子效应能有效促进ISC,但是限于经济问题和物质储量,制备无稀有金属参与的光敏剂提上了日程。相对原子质量较大的碘、溴等元素也具有很强的重原子效应,可以实现高效的ISC,而且对于三重态寿命的削弱作用相对较弱[52]。在BODIPY的2,6位引入碘原子可以有效的增强光敏剂的ISC。图12所示的化合物均具有高ISC产率(如化合物3-14的ΦΔ的值为79%,三重态寿命为84.6 μs)[53],这对双碘取代BODIPY类光敏剂的光催化应用提供了保证。
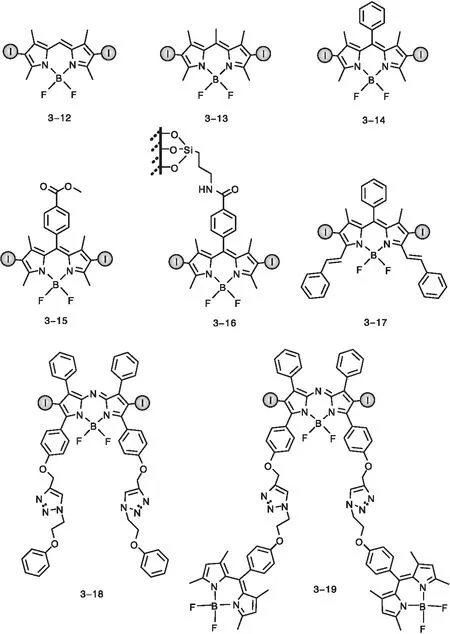
图12 无金属碘代BODIPY光敏剂的分子结构Figure 12 Structures of iodinated BODIPY derivatives containing no metal as photosensitizers
亚砜类物质在医药科学和不对称合成上有着重要的应用,其中一种高效制备亚砜类物质的方法是利用相应的硫醚化合物,经光氧化过程实现亚砜类物质的制备[35, 54]。硫醚光催化氧化制备亚砜类化合物的机理如图13所示,其中光敏剂受光激发后产生的具有强氧化性的单线态氧是催化氧化的核心物质[16]。Li W等[55]将具有高单线态氧量子产率的光敏剂3-12~3-14应用于光催化氧化硫醚的反应中,这3种光敏剂的ΦΔ值分别为81%、64%和83%,且吸光能力强(摩尔消光系数最高可达81 000 M-1·cm-1)。以甲醇做溶剂,苯甲硫醚为反应底物,分别以3-12~3-14作为光敏剂进行光催化氧化反应,其中3-12和3-14体系在3 h即可实现硫醚化合物的完全转化,证明碘代BODIPY光敏剂在此类反应中的高效光催化活性。

图13 硫醚光催化氧化制备亚砜类化合物的机理Figure 13 Schematic representation of photocatalytic oxidation of thioanisole to phenyl sulphoxide
为了利用红光波段的能量作为激发能量源,需要延长光敏剂的吸收至红光区。延长化合物3-14的共轭体系可得到光敏剂3-17,其最大吸收波长为630 nm,且ΦΔ的值为69%。赵建章教授课题组[53]将光敏剂3-14与3-17分别应用于苄胺的氧化偶联反应以及一锅法氧化萘酚制N-苯胺取代萘醌反应(图14),其中苄胺的氧化偶联反应为单电子转移机理,在物质的量分数为1%的3-14和室温光照1 h条件下,可以实现较高的反应产率(苄胺为底物时的产率大于99%)。而在萘酚氧化体系中加入物质的量分数为10%的乙酸铜作为催化剂,可以实现N-苯胺取代萘醌的一锅法合成。

图14 (a)苄胺的氧化偶联反应;(b)氧化萘酚制N-苯胺取代萘醌的催化机理[53]Figure 14 Schematic representation of (a) photocatalytic aerobic oxidative coupling of amines and (b) photocatalytic oxidative of naphthol to N-aromatic amines substituted naphthoquinone[53]

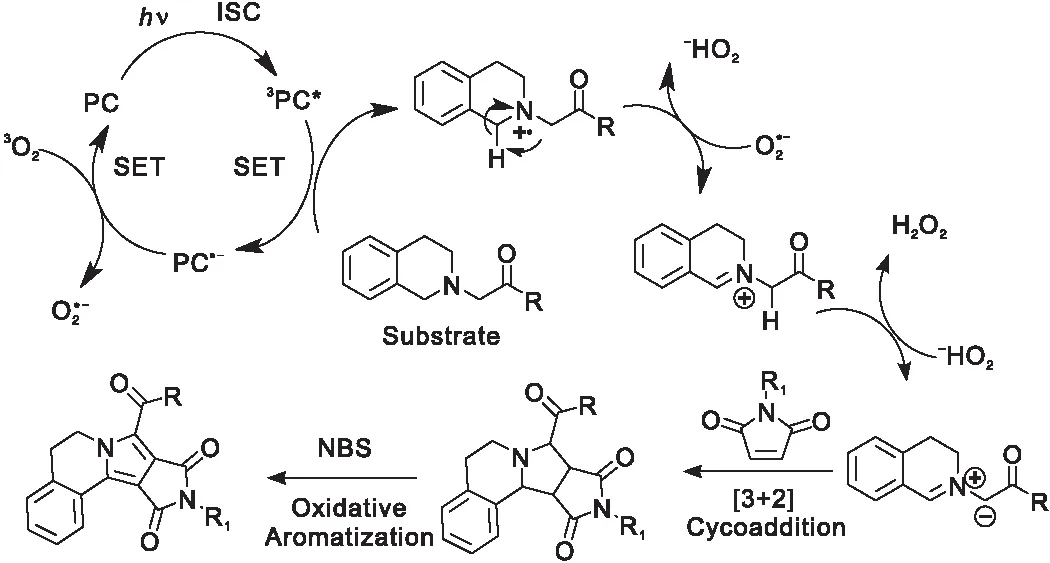
图15 光催化氧化-[3+2]环加成-芳构化串联反应机理[58]Figure 15 Schematic representation of photocatalytic oxidation-[3+2]cycloaddition-aromatization cascade reaction[58]
赵建章教授课题组将光敏剂3-14~3-19应用于催化该类反应中[56-58],光敏剂3-14、3-17与传统商用的光催化剂1-1相比,催化反应时间从(11~24) h缩减到1 h,TON值也有相应的提高[56,59-60]。相对于非均相催化剂,均相光催化剂很难从反应混合物中分离、回收再利用继续进行催化反应,因此该课题组将光敏剂3-14负载到硅质介孔分子筛KIT-1上,获得非均相催化剂3-16,并用于催化吡咯类异喹啉的制备[58]。实验证明,与均相催化剂相比,负载后的催化剂3-16的催化活性并没有明显下降,以日光为光源的光催化效率为光催化剂1-1的3倍。
为提高对可见光的利用率,该课题组设计并合成了光敏剂3-18与3-19[57],引入吸收波长更长的碘代aza-BODIPY作为催化核心发色团,非共轭连接两个BODIPY吸光基团,将吸收波长延长至700 nm,可有效利用红光与近红外光的能量。其中,光敏剂3-19的吸收光谱覆盖了(400~700) nm的全可见光区,利用荧光共振能量转移(FRET)机理实现了对白光催化光源的有效利用。
原子转移自由基加成反应(ATRA)是以烯烃与卤代物为反应底物进行的自由基加成反应,获得碳-碳或碳-卤键连接的功能化产物,该反应已经发展成为一种具有高原子经济性和有效的直接功能化烯烃底物的方法(图16)[61]。Magagnano G和Lama A D等[62-63]利用光催化手段,以苯甲酮类化合物作为光敏剂实现了高效的原子转移自由基加成反应,将具有长波长强吸收的光敏剂3-14引入原子转移自由基加成光催化反应中,代替弱吸收短波长的苯甲酮类物质,也可以实现高效的底物转化。
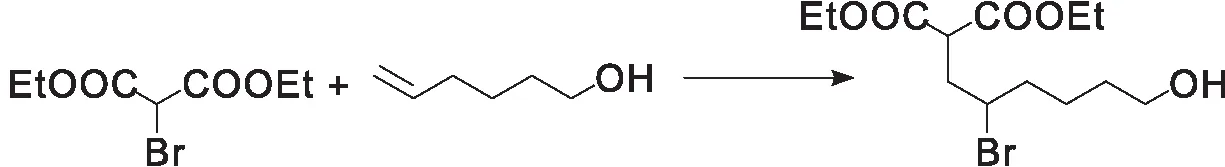
图16 原子转移自由基加成反应(ATRA)[62]Figure 16 Schematic representation of atom-transfer radical addition (ATRA)[62]
溴原子也具有重原子效应,因此溴代BODIPY类光敏剂也可实现光催化的过程。Wang X F等[64]利用光敏剂3-20作为光催化剂(图17),在2,6-二叔丁基对甲酚(BHT)的存在下,实现了可见光激发的芳香醛酰胺化反应(图18a),以对溴苯甲醛和吡咯烷为底物,日光照射6 h后实现了90%的目标产物收率。García-Santos W H等[65]利用光敏剂3-21作为光催化剂(图17),以α-卤代羰基化合物和乙烯基醚类化合物为底物,绿光LED为光源,通过原子转移自由基加成反应直接制备γ-烷氧基内酯、单保护1,4-酮醛和二氢呋喃类物质(图18b)。Stafford A等[66]将化合物3-22和3-23作为光敏剂应用于光致3D打印领域(图17),实现了在极低的光强度(<1 mW·cm-2)和催化剂负载(<50 μM)条件下的高速聚合:分别以绿色、红色和远红外LED作为光源,照射60 s即可实现反应完全。尤其是以远红外LED为光源实现高分辨率的3D打印,更加具有应用价值。

图17 无金属溴代BODIPY光敏剂的分子结构Figure 17 Structures of brominated BODIPY derivatives containing no metal as photosensitizers
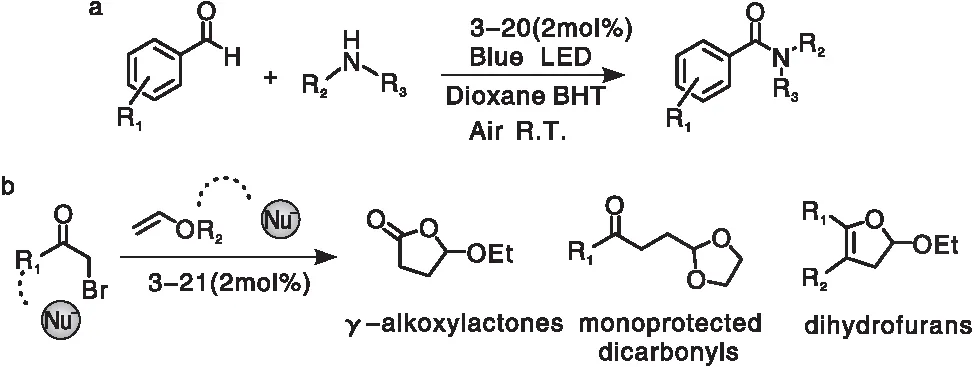
图18 (a) 3-20催化的芳香醛酰胺化反应;(b) 3-21催化的原子转移自由基加成反应[64-65]Figure 18 Schematic representation of (a) amidation of aromatic aldehydes with 3-20 as photocatalysts and (b) atom-transfer radical addition with 3-21 as photocatalysts[64-65]
光解水产氢是光催化的一个很重要的研究方向[67]。以碘代BODIPY类化合物为光敏剂,金属或金属配合物为催化剂可以应用于光解水产氢领域(图19)。2011年,Eisenberg R课题组[68]首次将光敏剂3-15应用于光催化产氢反应中,以Pt/TiO2为催化剂、三乙醇胺(TEOA)为电子牺牲试剂,该体系可以在氙灯的光照下稳定催化产氢20 h。并且作者发现,溴代BODIPY光敏剂的产氢速率要高于3-15,但是TON值仅为3-15的一半,而且光稳定性较差,仅能维持(3~4) h。骆耿耿课题组[69]将化合物3-24和3-25作为光敏剂应用于光催化产氢反应中,当以化合物3-26为催化剂,TEOA为电子牺牲试剂时,可以实现有效的光解水产氢,作者还发现邻位羧基取代的光敏剂3-25催化效率高于对位取代的光敏剂3-24(3-25的TON值为197,3-24的TON值为126)。
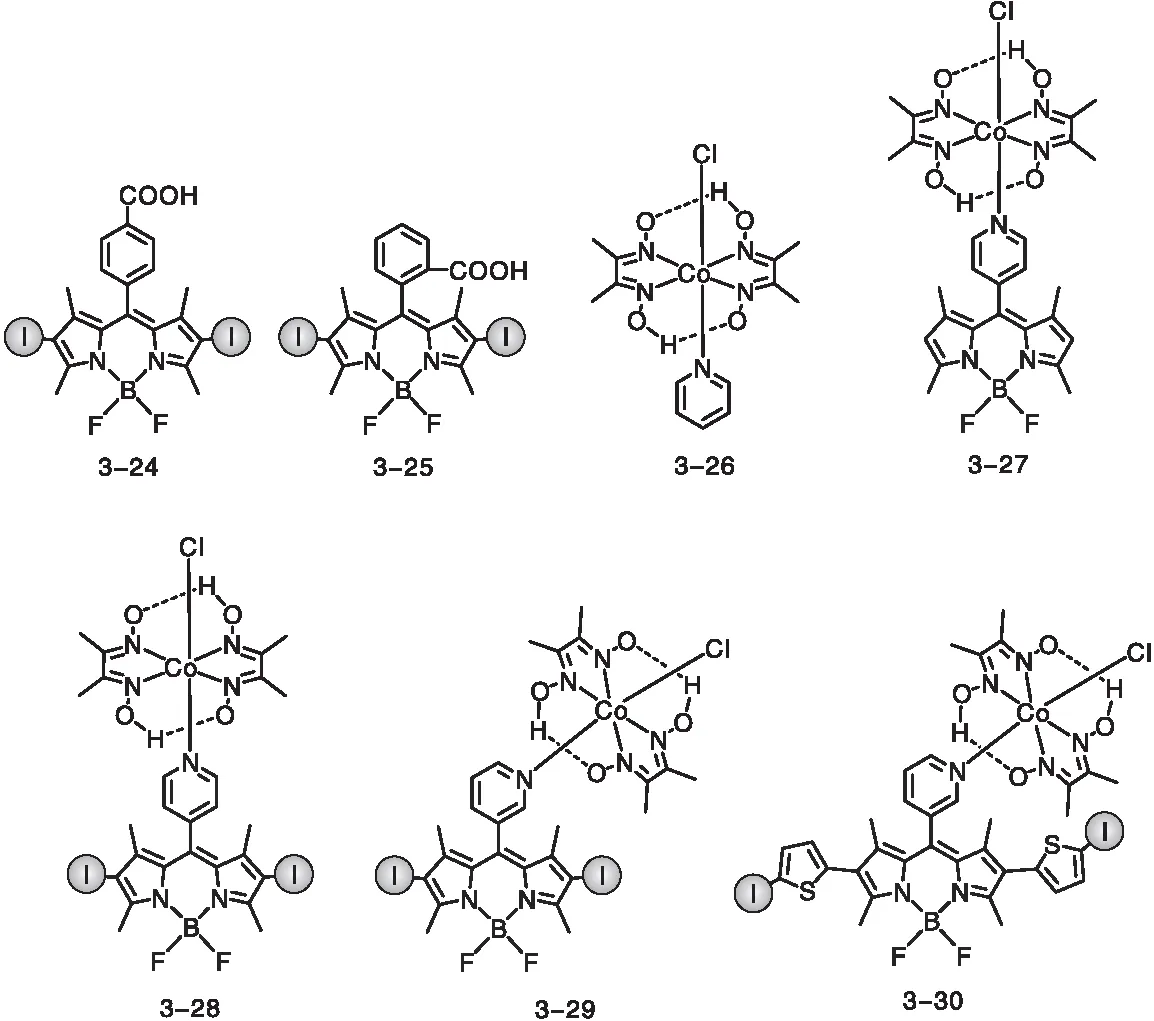
图19 应用于光解水产氢的碘代BODIPY光敏剂的分子结构Figure 19 Molecular structures of iodinated BODIPY derivatives for photocatalytic H2 evolution
上述光催化反应均基于光致电子转移机理,即光敏剂的激发态与作为催化剂的金属配合物发生电子转移,引发进一步的电子链传递反应。而多组分的催化体系中,光敏剂与催化剂发生的是分子间电子转移,其电子转移速率通常慢于分子内电子转移过程。因此将光敏剂以配位的形式与中心金属直接配位,可以提高光催化产氢的效率。Bartelmess J等[70]将meso位吡啶取代的碘代BODIPY光敏剂与钴肟配合物进行配位得到光催化剂3-28,并将其应用于光解水产氢实验中,发现直接配位的光催化剂3-28的光稳定性和产氢效率均高于相应条件下[光敏剂+催化剂]模式的二元催化体系。由于在二元催化体系中,BODIPY结构中取代基的邻位效应对反应有明显的影响,所以Luo G G等[11]进一步研究了一元催化体系中钴肟配合物与碘代BODIPY基团的相对取代位置对光解水产氢效率的影响,实验发现对于一元催化体系,发色团与金属配合物的邻位效应对于产氢效率并无明显影响。Luo G G等[71]又将噻吩引入BODIPY的2,6位获得光催化剂3-30,该策略不仅延长了光敏剂的吸收波长,还提高了催化剂的稳定性。与多组分催化体系相比,在相应的条件下,使用光催化剂3-30的TON值提升了2.5倍,催化效率提高了3倍。
金属有机框架(MOF)材料是一种三维多孔材料,具有比表面积大、孔隙率高以及孔径可调等特点,在催化领域有着广泛的应用[72]。将碘或溴原子取代的BODIPY光敏剂以共价键连接的方式负载到MOF上,可以对一些反应进行光催化[73-76]。Quan Y等[73]将双碘BODIPY共价连接到Zr-UiO-68材料上(如图20a),实现了氧气条件下的脱氢交叉偶联反应以及[3+2]环加成反应的光催化。

图20 负载BODIPY发色团的MOF材料结构示意图[73-74]Figure 20 Schematic representation of immobilized BODIPY based metal-organic framework (MOF)[73-74]
Atilgan A等[74]将双溴BODIPY共价连接到NU-1000材料中(如图20b),利用光敏剂受光激发产生的单线态氧,实现了高毒性物质芥子气及其衍生物的光催化氧化,将硫醚类物质氧化为砜类物质(如图11所示)。
2.2.3 无重原子的BODIPY类光敏剂
上述的光敏剂主要利用了稀有金属或者溴、碘等原子的重原子效应实现高效的ISC,但是存在稀有金属价格高储量小、溴或碘取代的化合物具有生物环境毒性较高等缺点,且光稳定性相对较差。为了避免这些不利影响,可以通过一些设计策略制备无重原子的三重态光敏剂[43,77]。
利用物质本身高效的ISC能力,使其作为自旋转换单元(将自旋为单重态的分子转换为三重态),并修饰吸光能力强的发色团作为光吸收天线,结合二者的优势,可以制备具有强吸收和高效ISC的光敏剂。富勒烯(C60)的ISC效率接近100%[78],但其吸光能力很弱,不宜单独作为光敏剂使用。因此基于上述机理,赵建章教授课题组报道了BODIPY-C60化合物并应用于光催化反应(如图21)。C60在最大吸收波长处的摩尔消光系数仅为6 500 M-1·cm-1,而经修饰后的光敏剂3-31和3-32的摩尔消光系数分别为76 000 M-1·cm-1和64 000 M-1·cm-1,ΦΔ的值分别为81%和82%,显示出该策略设计的光敏剂具有成为光催化剂的潜力[79]。作者将光敏剂3-31和3-32作为光催化剂进行[3+2]环加成反应,在添加物质的量分数1%光敏剂的条件下,光照1 h后反应产物的产率约达到90%;作为对照反应的C60和化合物1-1在相应条件下,光照4 h的产率仅分别为17%和37%。为了实现具有宽谱带吸收的光催化剂,作者进一步设计合成了化合物3-33,该化合物的吸收谱带可以覆盖(400~700) nm的波长范围。将3-33作为光催化剂应用到1,5-二羟基萘氧化制胡桃醌以及芳基硼酸催化氧化制酚的反应中,取得了很好的催化效果[80]。BODIPY-C60类化合物在这两个反应的光催化机理分别为能量转移的单线态氧机理和单电子转移机理,表明此类光敏剂具备较广泛的光催化应用价值。

图21 引入自旋转换单元(C60)的三重态光敏剂Figure 21 Triplet photosensitizers based on C60 with spin-crossover effect
电荷分离再复合也可以促进ISC的产生,即分子受光激发后到达单重激发态,进而发生电荷转移,最后通过电荷复合获得三重激发态[81-83]。电荷分离体系中,可以将电子给体(D)和电子受体(A)直接相连,通过电荷复合直接生成定域在发色团上的三重激发态,这一机理被称为自旋轨道耦合电荷转移系间窜越(SOCT-ISC)[84]。由于此类化合物的分子结构简单、易于合成、不含有重原子且生物毒性低,可应用于光动力治疗领域[36,85]。基于SOCT-ISC机理光敏剂的ISC效率较高,近年来也被应用于光催化领域[86-87]。Cakmak Y等[88]在2011年发现了两个BODIPY发色团在meso位直接相连的化合物3-34具有较高的ISC效率(氯仿中ΦΔ的值为46%),并将其应用于光动力治疗领域(见图22)。Wang L等[89]在2014年报道的文章中发现,化合物3-34在高极性溶剂中荧光被明显淬灭,证明具有电荷转移的性质,将其作为光催化剂应用于硫醚光催化氧化制备亚砜类的反应中,取得较好的催化效果,但未指出该化合物ISC的具体机理。Abuhadba S等[90]于2021年报道了以BODIPY-AN化合物为光催化剂的ATRA反应体系(图22,3-35),证明了基于SOCT-ISC机理的光敏剂光催化能力。

图22 基于SOCT-ISC机理的三重态光敏剂Figure 22 Structures of triplet photosensitizers based on SOCT-ISC
张志明和郭颂等[87]于2022年报道了一系列光敏剂(图22,3-36~3-38),该系列光敏剂的吸收波长可以覆盖(450~670) nm的光谱谱带,作者以化合物3-38为光敏剂,镍盐为催化剂,抗坏血酸为电子牺牲试剂,氙灯中大于600 nm波段的光为光源,实现了可观的光解水产氢效果。这一体系实现了以红光和红外光激发的光解水产氢,拓展了无重原子三重态光敏剂的应用范围,开拓了对红光和红外光的利用。
3 结论与展望
经济社会的可持续发展使得新能源的开发与利用必然会日益广泛和深入,太阳能作为最佳的可持续利用能源,如何有效的利用光能转化为化学能,是实现化学合成节能化和可持续化发展的路径之一。光催化剂作为光催化化学合成体系的核心,如何设计出安全、经济、稳定、高效的光催化剂是推进光催化发展的前提条件。以BODIPY发色团为核心的三重态光敏剂广泛应用于光催化合成领域,此类新型光敏剂具备催化多种合成反应以及催化光解水产氢的能力。与传统的三联吡啶钌、三(2-苯基吡啶)铱等光催化剂相比,无论是将BODIPY发色团作为配体引入金属配合物还是直接重原子化,均能实现更加高效的光催化能力。BODIPY类三重态光敏剂具有可见光区的强吸收、长三重态寿命以及易于衍生化等特点,可以根据不同的反应类型进行模块化设计,而无重原子三重态光敏剂可以进一步避免贵金属和重卤素原子的引入,从而节约成本和避免生态毒性。
目前对于BODIPY类的三重态光敏剂仍然存在着一些需要解决的问题:(1)相对于非均相催化剂,均相有机催化剂的回收再利用始终是一项挑战,如何能够方便快捷的对光催化剂进行回收并不失活性的再利用是一个需要持续研究的课题;(2)虽然均相有机光催化剂的反应活性和选择性很高,但是相对于无机非均相催化剂,如负载型分子筛,有机均相催化剂的稳定性仍然有很大需要提升的空间;(3)目前很多用于光催化的光敏剂的合成路线仍然较长,如何简便高效的合成光敏剂仍然需要探索;(4)需要继续探索新型三重态光敏剂的设计策略,进一步拓展其可催化的反应类型,从而扩展应用范围。
随着对光敏剂在光催化领域应用的逐渐深入,对上述困难与挑战的逐项优化将会对光化学和光催化领域的发展提供宝贵的理论基础和实验经验。
猜你喜欢
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
陶瓷学报(2019年6期)2019-10-27 01:18:18
山东化工(2019年2期)2019-02-16 12:38:10
福建农林大学学报(自然科学版)(2018年5期)2018-10-11 08:05:32
浙江农业科学(2016年11期)2016-05-04 04:16:45
原子与分子物理学报(2015年3期)2015-11-24 12:49:36
应用化工(2014年11期)2014-08-16 15:59:13
应用化工(2014年8期)2014-08-08 13:11:39
中国药理学通报(2014年2期)2014-05-09 08:22:16
原子与分子物理学报(2014年1期)2014-03-20 08:16:14
