
川渝大豆生育期性状的全基因组关联分析
2023-08-24 12:56:46向仕华宁可君舒英杰何庆元
作物学报 2023年10期
杨 豪 向仕华 刘 丽 宁可君 杨 雪 舒英杰 何庆元,*
川渝大豆生育期性状的全基因组关联分析
杨 豪1,**向仕华2,**刘 丽1宁可君1杨 雪1舒英杰1何庆元1,*
1安徽科技学院, 安徽凤阳 233100;2自贡市农业科学研究院, 四川自贡 643000
生育期是大豆品种适应生态环境进化的结果, 对产量和农艺性状都有重要影响。解析南方, 特别是挖掘川渝地区大豆生育期相关遗传位点并筛选出候选基因, 能为该地区大豆育种奠定一定的理论基础。以川渝地区227份大豆品种或资源为试验材料, 利用135个SSR标记和107,081个有效SNPs标记进行基因分型, 分别考察及统计了2016年安徽凤阳和四川自贡、2018年安徽凤阳3个环境的营养生长期、生殖生长期、全生育期以及营养生长/生殖生长期比值。川渝大豆4个性状的表型分布整体呈正态分布, 遗传变异受品种、环境和环境互作的显著影响。通过全基因关联分析, 使用SSR标记共检测到51个与生育期性状显著关联的位点, 使用SNP标记检测到70个与生育期性状显著关联的位点。其中在2个以上的环境下同时被检测的区域有: 位于13号染色体BLK_29175719-29275719和BLK_30878620_30978620, 14号染色体BLK_48763386_48863386以及16号染色体BLK_10093551_10293551区块, 在这4个区域内预测到11个与植物生长发育有关的可能候选基因, 同源基因预测表明6个与生育调控相关, 进一步的单倍型分析表明有3个基因单倍型在生育期性状表型上存在显著差异, 分别是、和。
大豆; 生育期; SNP; 全基因组关联分析
大豆((L.) Merr.)含有丰富的营养物质, 是最大植物蛋白质和第二大油料来源自花授粉作物[1]。大豆对光周期敏感, 生育期长短是大豆适应生态环境的结果, 对产量高低具有重要影响[2]。同时大豆生育期是一个复杂的数量性状, 受少数主效基因和一些微效基因共同控制[3]。到目前为止, 已经被证实存在与大豆开花和成熟有关的等位基因至少有13个, 分别是E1[4]、E2[5]、E3[6]、E4[7]、E5[8]、E6[9]、E7[10]、E8[11]、E9[12]、E10[13]、J[14]、Tof11和Tof12[15], 除E5[8]、E6[9]、E7[10]、E8[11]外其余9个已被成功图位克隆和证实, 这些基因主要决定光周期的敏感性[11]。已有的研究表明, 随着大豆种植生态区的扩大, 不同生态区内可能存在有新的影响生育期长短的等位变异[12]。
鉴定和验证更多生育期相关的等位基因, 能够加快选育适应不同生态区高产大豆品种。川渝地区作为南方大豆主产区之一, 经长期的自然和人工选择, 同样可能存在新的生育期相关变异。本研究选用227份不同川渝大豆种质资源, 利用全基因组关联分析鉴定了4个生育期性状, 即营养生长期(Vp)、生殖生长期(Rp)、全生育期(Wgp)和生育期结构(Rp/ Vp)的遗传位点。挖掘可能调控大豆生育期性状的候选基因, 为解析生育期相关性状的遗传机制和产量稳定、适应性广的川渝大豆新品种的选育奠定基础。
1 材料与方法
1.1 供试材料
选用来源于川渝地区的227份大豆种质资源, 其中209份材料为川渝地区地方品种, 另外18份材料为自贡市农业科学研究所和南充市农业科学院所育成的品种(附表1)。
1.2 试验方法
1.2.1 田间试验及生育期调查 所有供试材料分别于2016年和2018年种植于安徽凤阳(16FY和18FY) (32°47′N和117°19′E) 2016年种植于四川自贡(29°33′N和104°55′E) (16ZG)。采用随机区组试验设计, 重复3次, 每区块行长2 m, 行距0.4 m, 每行20株, 正常田间管理。营养生长期(Vp)是指从大豆播种至初花期(第1朵花)的天数, 生殖生长期(Rp)是指从大豆初花期至完熟的天数, 全生育期(Wgp)是指从大豆播种至完熟的天数, 生育期结构(Rp/Vp)是生殖生长期/营养生长期的比值。
1.2.2 DNA提取与基因分型 使用改良CTAB法[16]从幼嫩的大豆叶片中提取基因组DNA。SSR基因分型: 从Soymap2图谱中[17]选择在20条染色体上分布基本均匀的135对SSR引物进行基因分型[18]。使用8%的丙烯酰胺凝胶电泳检测PCR产物, 0.1%的AgNO3进行银染, 人工读取带型。SNP基因分型: 使用大豆200K基因芯片(北京康普生生物科技有限公司)进行基因分型, SNP芯片的内参质控和变异校准方法按照芯片操作手册执行, 使用Plink软件对基因型原始数据进行质控, 去掉缺失比例高于0.1的位点, 去除最小等位基因频率(maf)小于0.05的位点, 获得有效的SNP标记。
1.2.3 表型统计与分析 使用Microsoft Excel 2016整理大豆营养生长期、生殖生长期、全生育期和生育期结构。使用SPSS statistics 26进行数据处理, 主要包括数据的方差分析、变幅的统计以及变异系数和广义遗传率的计算。广义遗传率(2) = (遗传方差/总方差)。
1.2.4 全基因组关联分析 使用TASSEL 2.1和TASSEL 5.0软件基于混合线性模型(mixed linear models , MLM)、结合群体结构(Q)和亲缘关系(K) (Q+K), 分别进行生育期性状与SSR和SNP位点之间的全基因组关联分析。SSR的值分别设定为=0.05, SNP设定为=0.0001, 当检测位点值≤设定的阈值时, 即认为该位点与生育期存在显著关联。本群体分为3个亚群, 群体结构情况详见He等[19]的分析结果。
1.2.5 候选基因的预测 将检测到的与生育期性状紧密关联的位点映射到各自的单倍型块上, 参考SoyBase网站(http://www.soybase.org/) Glyma.Wm82. a2v1基因组, 在候选区域内查找生育期性状的候选基因, 并根据候选基因的注释信息来筛选可能的候选基因, 利用Phytozome数据库(http://phytozome.jgi. doe.gov/)查找候选基因的表达量分析。
1.2.6 单倍型分析 对候选基因的非同义突变SNP进行分型, 根据候选基因的不同单倍型将种质资源分组, 统计不同单倍型的占比, 利用R对候选基因每种单倍型品种数超过总品种数的5%表型进行不同单倍型组间性状的差异显著性测定, 以确定每种单倍型对表型的影响。
2 结果与分析
2.1 大豆生育期的表型分析
生育期性状(Vp、Rp、Wgp和Rp/Vp)在3个环境(16FY、18FY和16ZG)中均表现出较大的表型变异。在3个环境下, Vp、Rp、Wgp和Rp/Vp的变化范围分别在14~83、15~100、65~159和0.246~3.071之间。峰度和偏度的绝对值范围在0.008~2.308之间, 基本符合正态分布特征(图1), 4个性状的广义遗传率(2)范围在40.20%~97.49%之间, 变异系数(GCV)范围在12.92%~27.17%之间, 遗传率在营养生长期中(16FY)最高, 在生育期结构中(18FY Rp/Vp)最低。变异系数(GCV)在生育期结构中(18FY Rp/Vp)最高, 在全育期(16ZG Wgp)最低(表1)。由各表型的方差分析和遗传率可知, 营养生长期、生殖生长期、全生育期和生育期结构显著受品系、环境和品系与环境的相互作用的影响。同时说明群体的生育期性状存在真实的遗传差异可以进一步进行关联分析(表2)。
2.2 生育期性状的全基因组关联分析
通过TASSEL 2.1软件进行关联分析, 3个环境下共检测51个与生育期相关的位点, 其中与营养生长期、生殖生长期、全生育期和生育期结构相关的位点分别有12、11、21和7个。有3个与营养生长期相关的位点在2个环境中同时被检测到, 其中Satt359与Satt553在16FY和16ZG, Satt129在16FY与18FY中被检测到。仅有1个与生殖生长期相关的位点(Sat_137)在2个环境中(16ZG和18FY)被检测到。在3个环境中都检测到的与全生育期相关的位点有3个, 分别是Sat_406、Satt332与Satt359, 同时有3个位点在16ZG和18FY两个环境中被同时检测到, 分别是Satt129、Satt553与Satt554。其余位点仅在单个环境中被检测到(表3)。
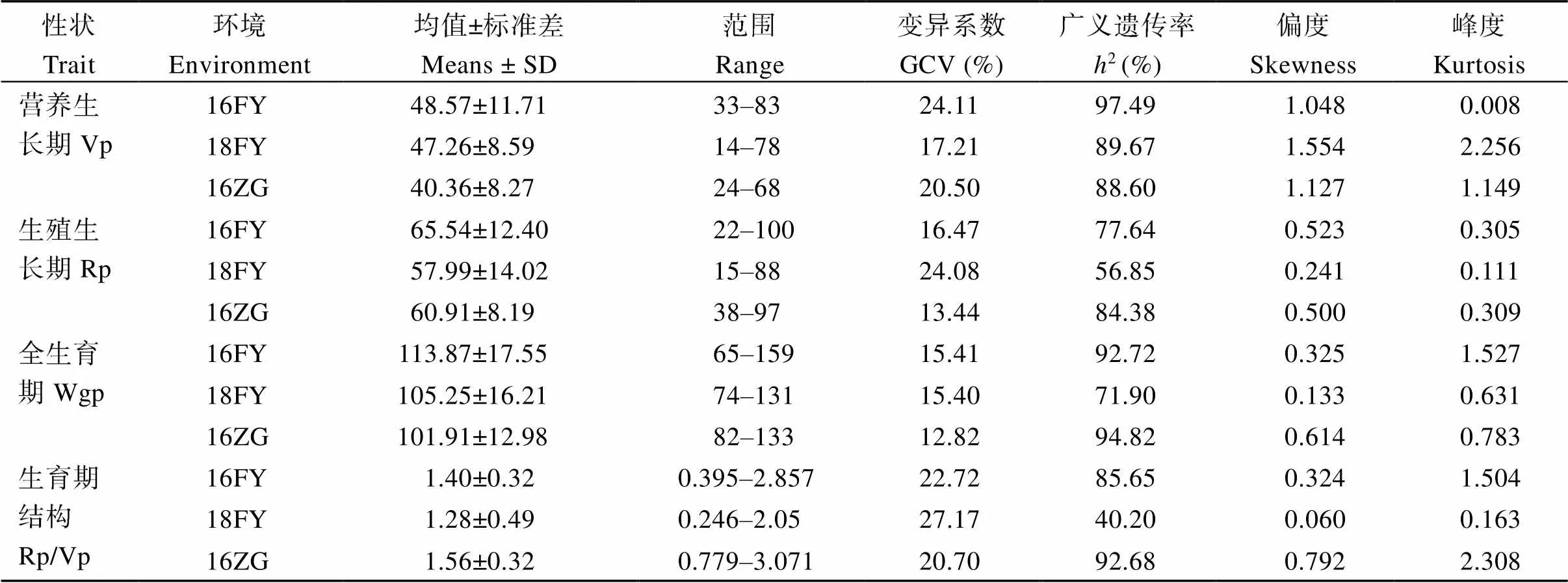
表1 3个环境下大豆生育期性状的表型分析
16FY: 2016年凤阳; 18FY: 2018年凤阳; 16ZG: 2016年自贡。
Vp: vegetative period; Rp: reproductive period; Wgp: whole growth period; Rp/Vp: reproductive period/vegetative period. 16FY: in 2016Fengyang; 18FY: in 2018 in Fengyang; 16ZG: in 2016 in Zigong.
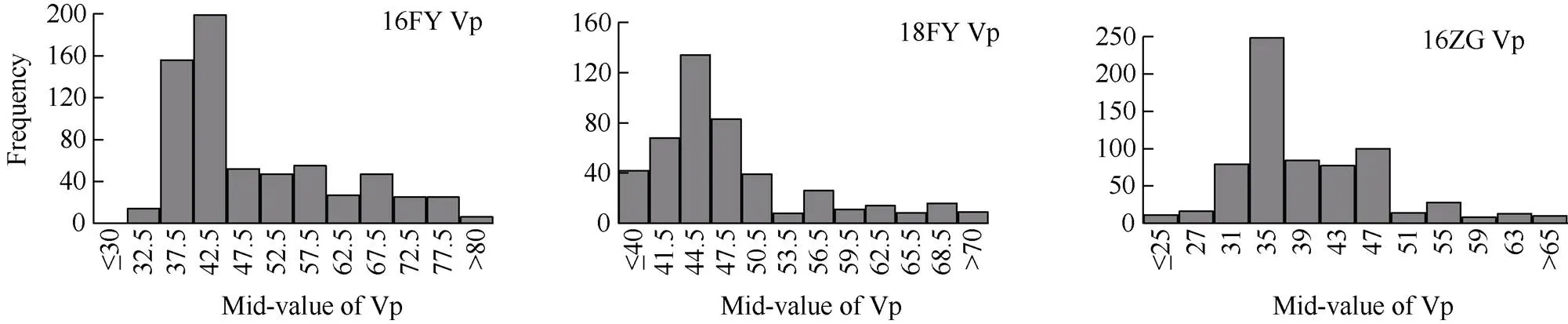
(图1)
Vp: 营养生长期; Rp: 生殖生长期; Wgp: 全生育期; Rp/Vp: 生育期结构。16FY: 2016年凤阳; 18FY: 2018年凤阳; 16ZG: 2016年自贡。
Vp: the vegetative period; Rp: the reproductive period; Wgp: whole growth period; Rp/Vp: the reproductive period/vegetative period. 16FY: in 2016 in Fengyang; 18FY: in 2018 in Fengyang; 16ZG: in 2016 in Zigong.
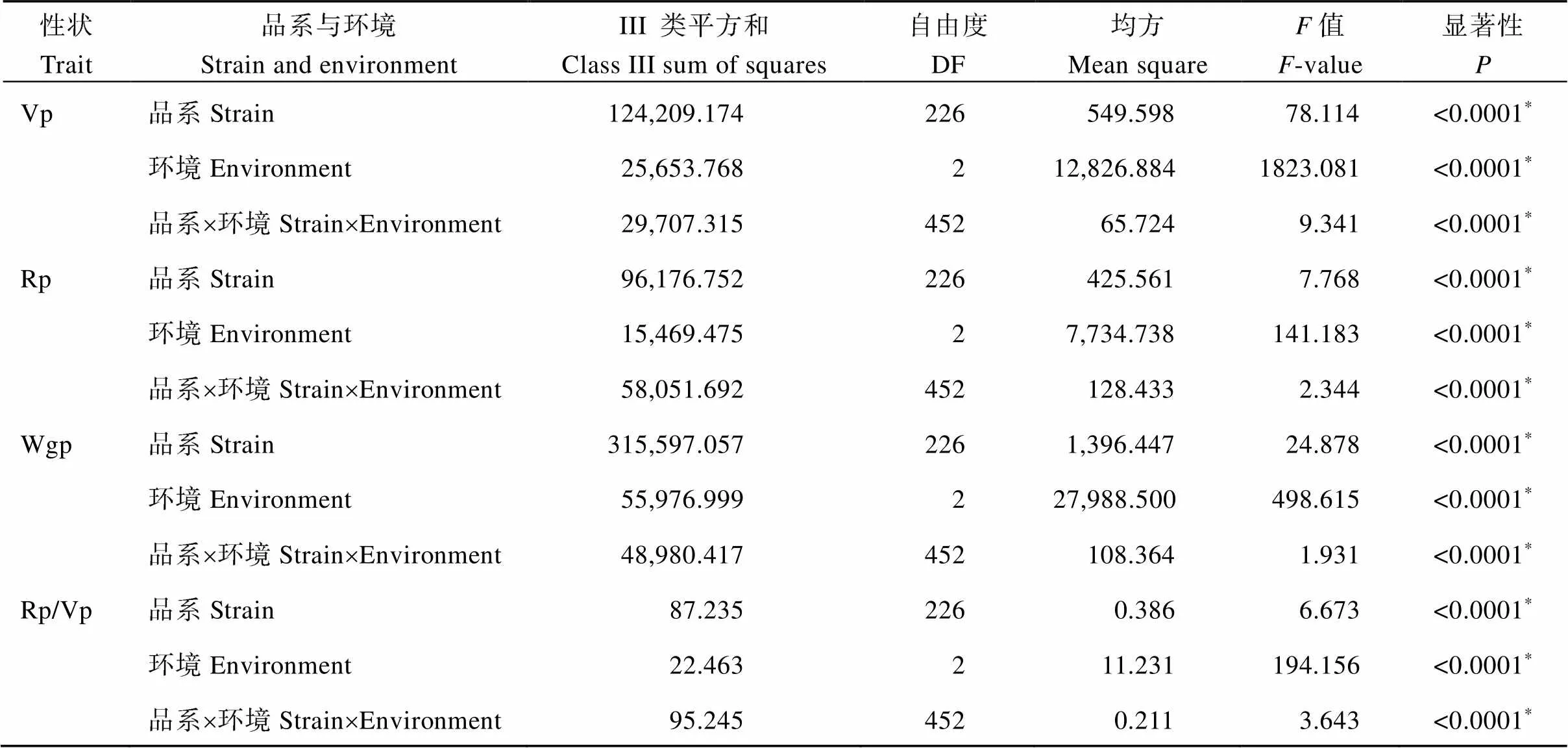
表2 大豆生育期性状的方差分析
Vp: 营养生长期; Rp: 生殖生长期; Wgp: 全生育期; Rp/Vp: 生育期结构。*:< 0.001。
Vp: the vegetative period; Rp: reproductive period; Wgp: the whole growth period; Rp/Vp: the reproductive period/vegetative period.*:< 0.001.

表3 在2个以上的环境中使用SSR标记与生育期性状相关的QTL
16FY: 2016年凤阳; 18FY: 2018年凤阳; 16ZG: 2016年自贡。
Vp: the vegetative period; Rp: reproductive period; Wgp: the whole growth period; Rp/Vp: the reproductive period/vegetative period. 16FY: in 2016 in Fengyang; 18FY: in 2018 in Fengyang; 16ZG: in 2016 in Zigong.
2.3 SNP对生育期的全基因组关联分析
使用Tassel 5.0软件对107,081个有效的单核苷酸多态性连锁(SNP)标记进行检测, 在混合线性模型(MLM)中, 共检测到70个具有统计学意义(=1×10-4有统计学意义)的SNPs位点(附表2)。
结果表明, 有51个SNP位点与营养生长期相关,分别位于5 (5个)、12 (9个)、13 (6个)、14 (1个)、16 (29个)和20 (1个)号染色体上, 可解释7.304%~ 14.144%的表型变异。7个SNP位点与全生育期相关, 分别位于8 (1个)、12 (1个)、13 (3个)、14 (1个)和16 (1个)号染色体上, 可解释7.522%~10.401%的表型变异。12个SNP位点与生育结构相关, 分别位于2 (1个)、3 (1个)、10 (2个)、11 (1个)、13 (5个)、17 (1个)和20 (1个)号染色体上, 可解释7.494%~ 12.848%的表型变异。相同位点在2个环境下检测到与生育期相关的SNP有3个, 分别是Gm13_ 29225719, 在3个环境中(16ZGWgp、16FYVp、16FYRp/Vp)的表型变异解释率为9.241%、10.944%和9.285%; Gm13_309286220在2个环境中(16ZGWgp、16FYVp)的表型变异解释率为8.572%和8.702%; Gm16_10193551在2个环境中(16ZGVp、18FYVp)的表型变异解释率为7.976%、9.909%; 有1个位点(Gm14_48813386)在16ZG同时检测到与Vp和Wgp相关联, 可解释7.81%~8.693%的表型变异(图2和表4)。
将SSR和SNP 2种定位结果映射到参考基因组(Glyma.Wm82.a2v1 )进行比对, 定位结果相近区域的有3个, 分别是位于5号染色体上SSR标记Satt648关联到的18FYWgp和SNP标记Gm5_32195936关联到的18FYVp, 位于14号上SSR标记Satt534关联到的18FYVp和SNP标记Gm_1448813386关联到的16ZGVp, 以及位于16号染色体上SSR标记Satt620关联到的16FYVp, 16FYWgp和SNP标记Gm16_21944024关联到的18FYVp (附表3)。

(图2)
(图2)
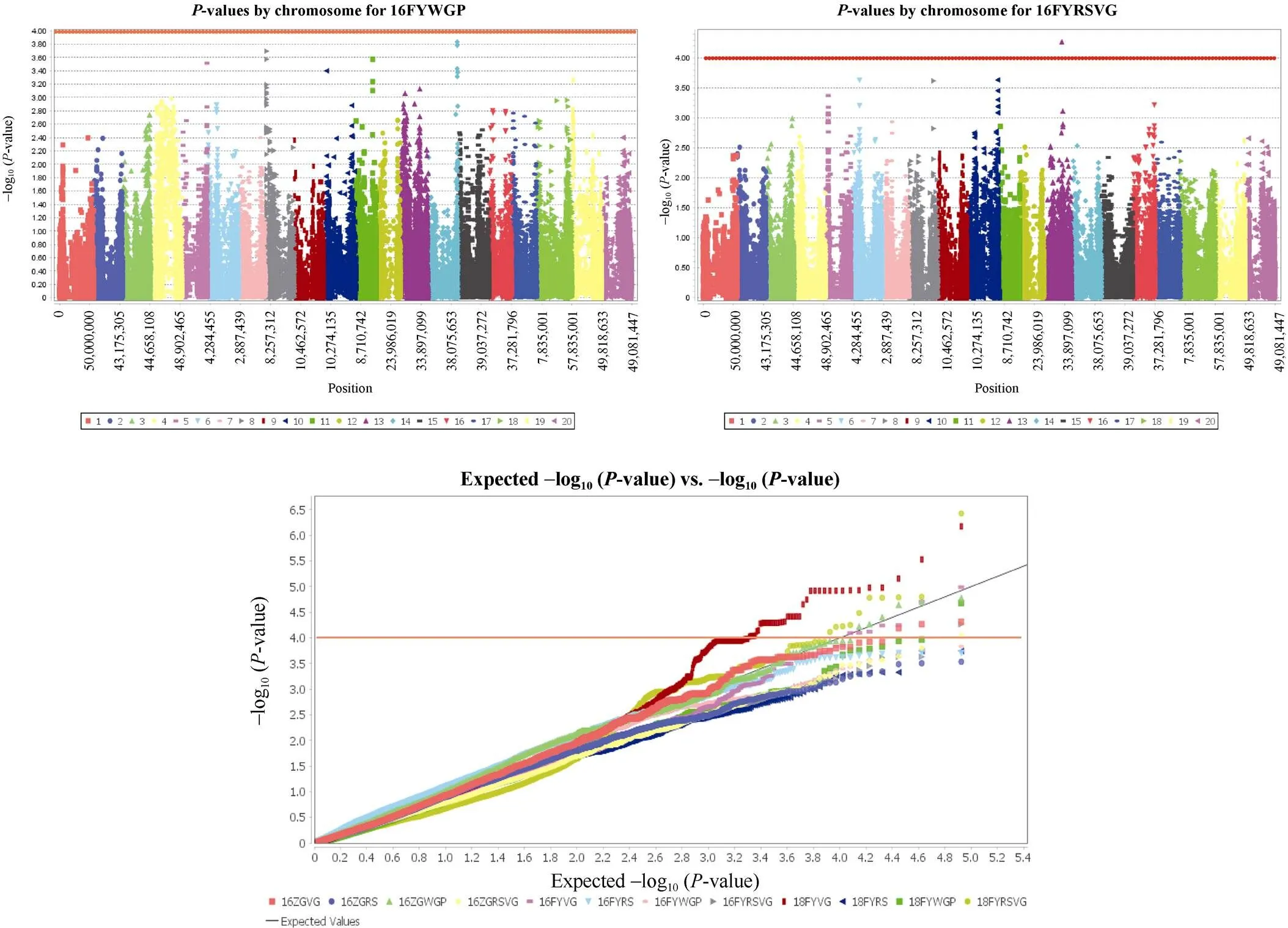
图2 大豆生育期相关性状全基因组关联分析曼哈顿图及QQ-Plot图
红线代表=1×10-4。6ZG: 2016年自贡; 18FY: 2018年凤阳; 16FY: 2016年凤阳; VG: 营养生长期; RS: 生殖生长期; WGP: 全生育期; RSVG: 生育期结构。
The red line represents=1×10-4. 16ZG: in 2016 in Zigong; 18FY: in 2018 in Fengyang; 16FY: in 2016 in Fengyang; VG: the vegetative period; RS: the reproductive period; WGP: whole growth period; RSVG: the reproductive period/vegetative period.
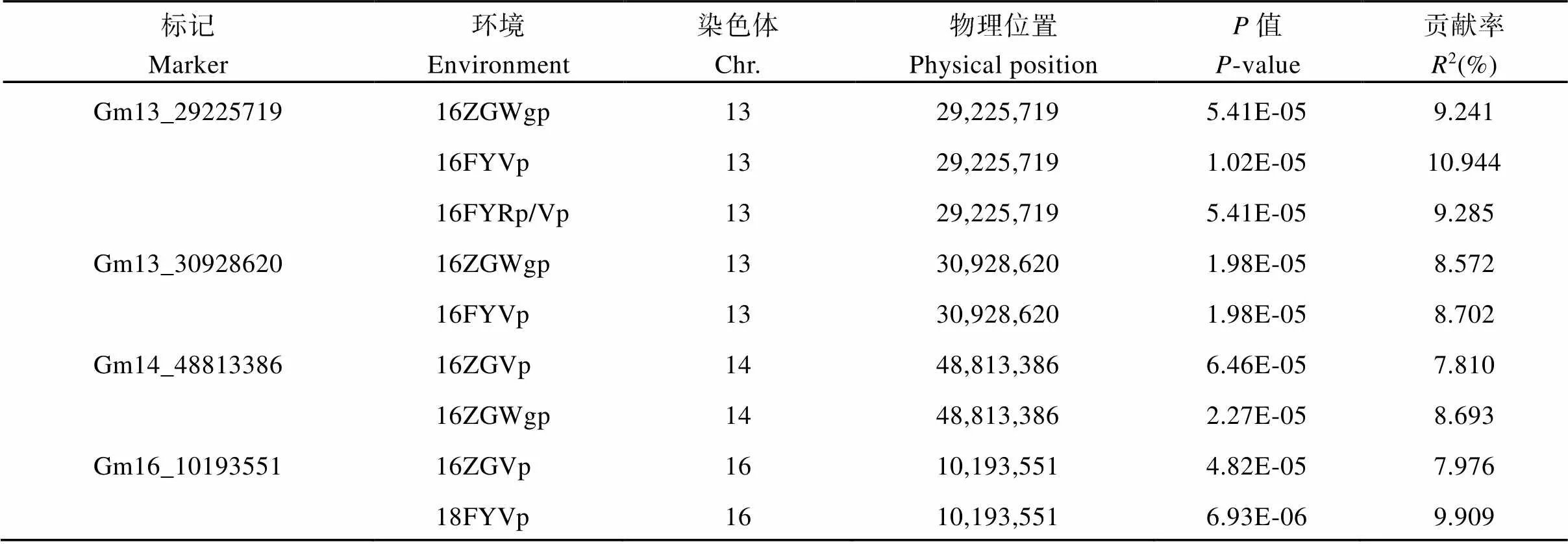
表4 使用SNPs在2个环境以上与生育期性状有关的QTNs
Vp: 营养生长期; Rp: 生殖生长期; Wgp: 全生育期; Rp/Vp: 生育期结构。16FY: 2016年凤阳; 18FY: 2018年凤阳; 16ZG: 2016年自贡。
Vp: the vegetative period; Rp: the reproductive period; Wgp: whole growth period; Rp/Vp: the reproductive period/vegetative period. 16FY: in 2016 in Fengyang; 18FY: in 2018 in Fengyang; 16ZG: in 2016 in Zigong.
2.4 候选基因的预测
为了预测与生育期性状相关的候选基因, 本研究选择了包含环境最多的4个Block进行候选基因的查找和筛选, 其中2个位于13号染色体, 分别是BLK_29175719_29275719, BLK_30878620_30978620;1个位于14号染色体, 是BLK_48763386_48863386以及一个位于16号染色体, 是BLK_10093551_ 10293551, 并在SoyBase网站(http://www.soybase. org/)内使用大豆Glyma.Wm82.a2v1 基因组作为参考进行筛选, 在4个Block内通过功能注释和文献比对初步筛选到候选基因28个(附表4), 其中11个与植物生长发育密切相关, 在这些基因中有10个基因在SoyBase上有不同组织上的表达量, 在不同发育阶段的种子中的表达量差异较大(附表5); 根据同源预测的结果, 与大豆生长发育相关的基因主要是Glyma.13g177400、Glyma.13g177600、Glyma. 13g177800、Glyma.13g178500、Glyma.13g195200和Glyma.14g223300 (表5)。
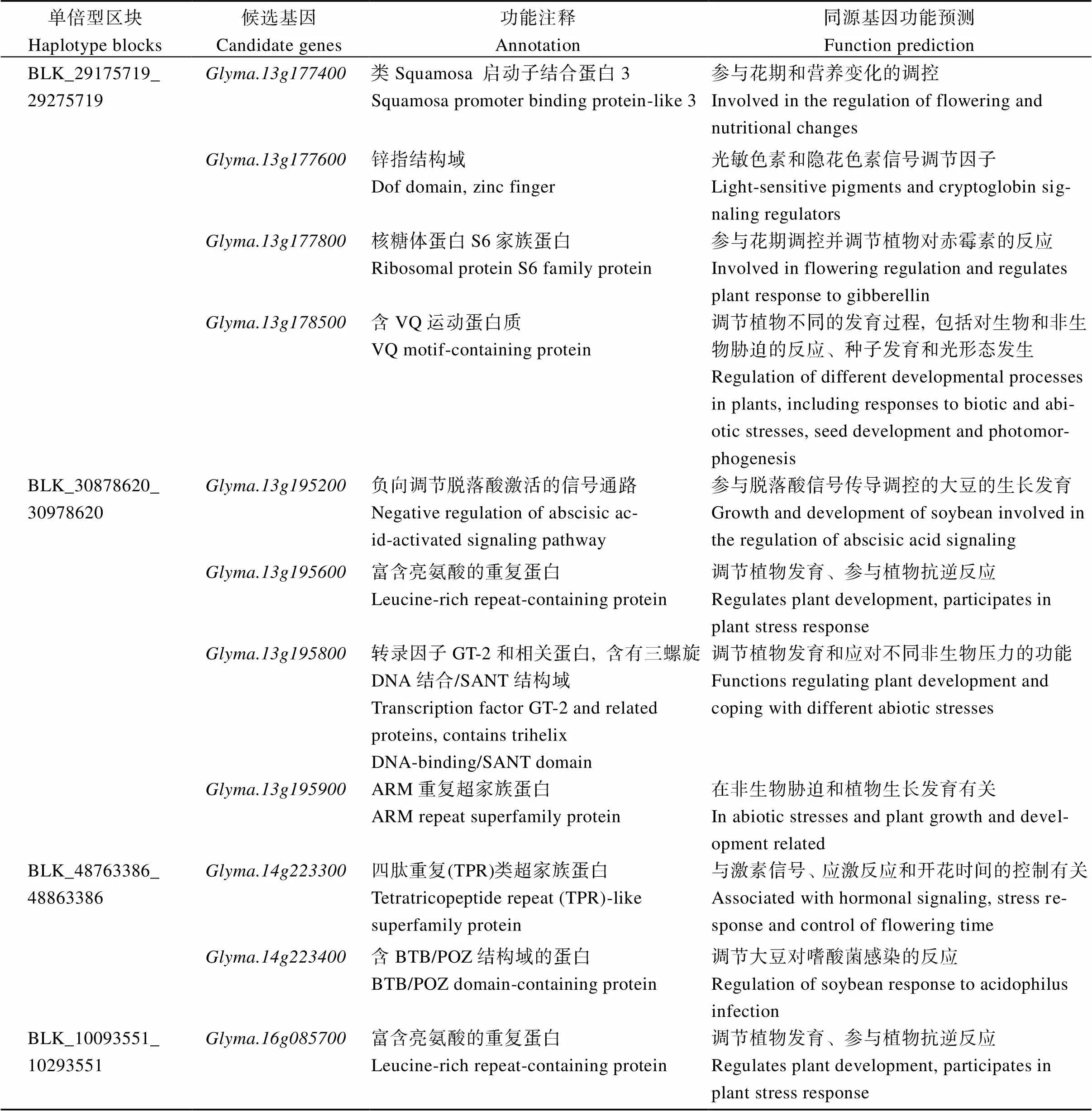
表5 与大豆生育期相关的候选基因及功能注释
2.5 候选基因的单倍型分析
对预测到的6个与生长发育有关的候选基因进行进一步的单倍型分析, 其中和基因内没有检测到SNP, 对区块两端与之相近的位点进行单倍型分析, 单倍型与之对应的表型性状之间没有显著性差异。被划分为2个单倍型, 但仅有一个单倍型数量大于5%, 无法通过资源群体验证该候选基因。
所在的Block关联到16FY环境下Vp和Rp/Vp存在显著性差异, 在16ZG的Wgp中没有显著差异, 被划分为2个单倍型, 在16FY环境中AA为晚花单倍型, 平均值分别为49.41 d, TT为早花单倍型, 平均值45.77 d; 在16FY环境中AA为生育期结构优异单倍型, 平均值为1.48, TT为生育期结构普通单倍型, 平均值为1.38。被划分为2个单倍型, 所在的Block关联到3个的性状(16FYVp, 16FYRp/Vp, 16ZGWgp)均存在显著性差异, 在16FY环境下AA为晚花单倍型和生育期结构一般单倍型, 平均值分别为50.40 d和1.35, GG为早花单倍型和生育期结构优异单倍型, 平均值分别为45.89 d和1.47; 在16ZG环境中AA为晚熟单倍型, 平均值为102.82 d, GG为早熟单倍型, 平均值为98.87 d。被划分为4个单倍型, 其中3个单倍型的数量大于5%, 所在的Block关联到的2个性状(16FYVp, 16ZGWgp), 但仅在16FYVp有显著性差异, 单倍型TTCC (早花单倍型)和TTTT (晚花单倍型)平均值分别为46.63 d和50.29 d (图3)。

图3 不同SNP类型材料对应的表型数据分析
3 讨论
大豆生育期性状与大豆产量有直接影响, 大豆的开花期和成熟期受基因和环境的共同影响[20]。本研究表明川渝地区大豆群体的营养生长期、生殖生长期、全生育期和生育期结构4个性状均受品系、环境和品系与环境的相互作用的显著影响, 与诸多研究结果一致, 大豆生育期受遗传和光周期的共同控制[21]。而在长期的进化过程中, 不同地区形成适应不同的生态类型, 在自然和人工的选择下形成新的等位变异。已有的研究表明通过对生育期结构进行定向选择, 可以提高大豆产量, 生殖生长期越长, 产量越高[22]。本研究使用SSR与SNP 2种标记进行全基因组关联分析, 分别定位到51个和70个位点, 分别有11个和4个位点在多个环境下被检测到。使用SNP定位到的位点并在多个环境下被检测到的位点中共有28个有注释功能的候选基因被检测到, 其中11个与植物生长发育密切相关, 候选基因预测到6个生育期相关的基因。
位于BLK_29175719_29275719内的基因与高度同源,与在功能上存在分化, 在大豆中主要参与株型的调控, 但不调控大豆营养生长期[23], 通过单倍型分析内没有检测到SNP的差异, 其原因可能是不存在差异单倍型或者本试验所用的SNP芯片密度不够, 有待进一步的候选基因测序分析。拟南芥DNA结合锌指(Dof)蛋白OBP3 (AT3G55370)为光敏色素和隐花色素下游信号传导成分之一,是Dof转录因子家族成员与拟南芥高度同源[24],的不同单倍型在表型上存在显著差异,推测可能参与光敏色素和隐花色素调控大豆生长发育的信号途径。是编码碱性螺旋-环形螺旋(bHLH) DNA结合超家族蛋白, 能够调节植物生长发育、植物抗逆和信息转导过程[25-26], 在拟南芥中主要参与花期调控并调节植物对赤霉素的反应[27], 但单倍型分析表明的表型没有显著差异。Glyma.13g178500是一类植物特异性VQ蛋白(VQ motif-containing protein), 可以调节植物不同的发育过程, 包括对生物和非生物胁迫的反应、种子发育和光形态发生[28]。拟南芥环型E3连接酶KEEP ON GOING (KEG, AT5G13530)是ABA信号转导的负调控因子。单倍型分析被划分为2个单倍型, 并在所关联的3个性状中均存在显著性差异, 其中AA单倍型是生育期结构优异单倍型。该Block内控制生育期的基因可能为和, 并且内存在为育种利用的优异等位变异。位于BLK_30878620_30978620内的与拟南芥高度同源, 推测其参与脱落酸信号传导调控的大豆的生长发育[29], 单倍型分析中TTCC (早花单倍型)和TTTT (晚花单倍型)在16FYVp有显著性差异。位于BLK_48763386_48863386内的, 这是一类四肽重复(TPR)类超家族蛋白, 主要与激素信号、应激反应和开花时间的控制有关[30], 但仅有一个单倍型数量大于5%, 无法通过资源群体验证该候选基因。
与生育期相关的5个候选基因进行单倍型分析的结果表明, 位于BLK_29175719_29275719内预测到3个与生长发育相关的基因, 其中的不同单倍型在表型上存在显著差异,没有显著差异, 可能定位到的结果是由所致。单倍型分析被划分为2个单倍型, 并在所关联的3个性状中均存在显著性差异, 其中AA单倍型是生育期结构优异单倍型; BLK_30878620_30978620内预测到1个候选基因,的2个单倍型(TTAA, TTTT)在16FYVp中显著差异, 关于大豆生育期的遗传机制仍有待进一步研究, 本研究检测到的候选基因将有助于对川渝地区大豆的生育期性状进行进一步了解。
4 结论
通过使用川渝地区的227份大豆品种, 使用SSR与SNP 2种标记在混合线性模型下行全基因组关联分析, 分别定位到51个和70个显著关联的标记。使用SSR标记共检测到12个与营养生长期相关的位点, 11个与生殖生长期相关的位点, 21个与全生育期相关及7个与生育期结构相关的位点。使用SNP标记共检测到51个与营养生长期相关的位点, 7个与全生育期及12个与生育期结构相关的位点, 未检测到与生殖生长期相关的位点。在多个环境下同时检测到的Block内有28个有注释功能的基因, 进一步的单倍体分析表明、和最有可能与大豆生育期性状相关。
附表 请见网络版: 1) 本刊网站http://zwxb. chinacrops.org/; 2) 中国知网http://www.cnki.net/; 3) 万方数据http://c.wanfangdata.com.cn/Periodical- zuowxb.aspx。
[1] Zhang Y H, Liu M F, He J B, Wang Y F, Xing N G, Li Y, Yang S P, Zhao T J, Gai J Y. Marker-assisted breeding for transgressive seed protein content soybean [(L.) Merr.]., 2015, 128: 1061–1072.
[2] Jia H C, Jiang B J, Wu C X, Lu W C, Hou W S, Sun S, Yan H R, Han T F. Maturity group classification and maturity locus genotyping of early-maturing soybean varieties from high-latitude cold regions., 2014, 9: e94139.
[3] Tasma I M, Shoemaker R C. Mapping flowering time gene homologs in soybean and their association with maturity (E) loci., 2003, 43: 319–328.
[4] Bernard R L. Two major genes for time of flowering and maturity in soybeans., 1971, 11: 242–244.
[5] Abe J, Xu D H, Miyano A, Komatsu K, Kanazawa A, Shimamoto Y. Photoperiod-insensitive Japanese soybean landraces differ at two maturity loci., 2003, 43: 1300–1304.
[6] Buzzell R I. Inheritance of a soybean flowering response to fluorescent-daylength conditions., 1971, 13: 703–707.
[7] Buzzell R I, Voldeng H D. Inheritance of insensitivity to long daylength., 1980, 7: 26–29.
[8] McBlain B A, Bernard R L. A new gene affecting the time of flowering and maturity in soybeans.,1987, 78: 160–162.
[9] Bonato E R, Vello N A. E6, a dominant gene conditioning early flowering and maturity in soybeans., 1999, 22: 229–232.
[10] Cober E R, Voldeng H D. A new soybean maturity and photoperiod-sensitivity locus linked to E1 and T., 2001, 41: 698–701.
[11] Cober E R, Molnar S J, Charette M, Harvey D. A new locus for early maturity in soybean., 2010, 50: 524–527.
[12] Zhao C, Takeshima R, Zhu J H, Xu M L, Sato M, Watanabe S, Kanazawa A, Liu B H, Kong F J, Yamada T, Abe J. A recessive allele for delayed flowering at the soybean maturity locus E9 is a leaky allele of FT2a, a FLOWERING LOCUS T ortholog., 2016, 16: 20.
[13] Samanfar B, Molnar S J, Charette M, Schoenrock A, Dehne F, Golshani A, Belzile F, Cober E R. Mapping and identification of a potential candidate gene for a novel maturity locus, E10, in soybean., 2017, 130: 377–390.
[14] Ray J D, Hinson K, Mankono J E B, Malo F M. Genetic control of a long-juvenile trait in soybean., 1995, 35: 1001–1006.
[15] Lin X, Liu B, Weller J L, Abe J, Kong F J. Molecular mechanisms for the photoperiodic regulation of flowering in soybean., 2021, 63: 981–994.
[16] Doyle J J. Isolation of plant DNA from fresh tissue., 1990, 12: 13–15.
[17] Song Q J, Marek L F, Shoemaker R C, Lark K G, Concibido V C, Delannay X, Specht J E, Cregan P B. A new integrated genetic linkage map of the soybean., 2004, 109: 122–128.
[18] He Q Y, Yang H Y, Xiang S H, Tian D, Wang W B, Zhao T J, Gai J Y. Fine mapping of the genetic locus L1 conferring black pods using a chromosome segment substitution line population of soybean., 2015, 134: 437–445.
[19] He Q Y, Xiang S H, Yang H W, Wang W B, Shu Y J, Li Z P, Yang X Y, Wang S H. A genome-wide association study of seed size, protein content, and oil content using a natural population of Sichuan and Chongqing soybean., 2021, 217: 198.
[20] Tasma I M, Lorenzen L L, Green D E, Shoemaker R C. Mapping genetic loci for flowering time, maturity, and photoperiod insensitivity in soybean., 2001, 8: 25–35.
[21] Li M M, Liu Y, Tao Y H, Xu C J, Li X, Zhang X M, Han Y P, Yang X, Sun J Z, Li W B, Li D M, Zhao X, Zhao X. Identification of genetic loci and candidate genes related to soybean flowering through genome wide association study., 2019, 20: 987.
[22] 杨倩, 张惠君, 谢甫绨. 不同来源大豆品种生育期结构与产量关系的研究. 大豆科学, 2008, 27: 973–978. Yang Q, Zhang H J, Xie F T. Study on the relationship between fertility structure and yield of soybean varieties of different origins.,2008, 27: 973–978 (in Chinese with English abstract).
[23] 吴艳, 侯智红, 程群, 董利东, 卢思佳, 南海洋, 甘卓然, 林永波. 大豆基因家族功能初探. 大豆科学, 2019, 38: 694–703. Wu Y, Hou Z H, Cheng Q, Dong L D, Lu S J, Nan H Y, Gan Z R, Lin Y B. A preliminary investigation of the function of thegene family in soybean.,, 38: 694–703 (in Chinese with English abstract).
[24] Ward J M, Cufr C A, Neff D M M. The Dof transcription factor OBP3 modulates phytochrome and cryptochrome signaling in.,2005, 17: 475–485.
[25] Dong Y, Wang C P, Han X, Tang S, Liu S, Xia X L, Yin W L. A novel bHLH transcription factor PebHLH35 fromconfers drought tolerance through regulating stomatal development, photosynthesis and growth in., 2014, 450: 453–458.
[26] Zhao Q, Xiang X H, Liu D, Yang A G, Wang Y Y. Tobacco transcription factor NtbHLH123 confers tolerance to cold stress by regulating the NtCBF pathway and reactive oxygen species homeostasis., 2018, 9: 381.
[27] Zhu Z G, Liang H L, Chen G P, Li F F, Wang Y S, Liao C G, Hu Z L. The bHLH transcription factor SlPRE2 regulates tomato fruit development and modulates plant response to gibberellin., 2019, 38: 1053–1064.
[28] Jing Y, Lin R. The VQ motif-containing protein family of plant-specific transcriptional regulators., 2015, 169: 371–378.
[29] Jiménez-López D, Muñóz-Belman F, González-Prieto J M, Victor Aguilar-Hernández, Plinio G. Repertoire of plant RING E3 ubiquitin ligases revisited: new groups counting gene families and single genes., 2018, 13: e0203442.
[30] Causier B, Ashworth M, Guo W, Davies B. The TOPLESS interactome: a framework for gene repression in., 2012, 158: 423–438.
Genome-wide association analysis of growth period traits in soybean of Sichuan and Chongqing
YANG Hao1,**, XIANG Shi-Hua2,**, LIU Li1, NING Ke-Jun1, YANG Xue1, SHU Ying-Jie1, and HE Qing-Yuan1,*
1Anhui Science and Technology University, Fengyang 233100, Anhui, China;2Zigong Institute of Agricultural Science, Zigong 643000, Sichuan, China
Growth period is the evolution of soybean varieties adaptded to the ecological environment. Which has important effects on the yield and agronomic traits. To lay the foundation for soybean breeding in southern China, especially in Sichuan andChongqing region.the relevant genetic loci of soybean growth period were analyzed and and candidate genes were screened out 227 soybean cultivars or resources from Sichuan and Chongqing regions were identified based on 135 SSR and 107,081 effective single nucleotide polymorphism (SNP) markers. Four growth period traits (vegetative growth, reproductive growth, whole growth and the ratio of vegetative growth / reproductive growth period) were investigated in three environments (Fengyang Anhui in 2016, Zigong Sichuan in 2016,and Fengyang Anhui in 2018).The results showed that the phenotypic variation of four traits followed normal distribution, and the genetic variation was significantly affected by variety, environment and environment interaction. A total of 51 loci and 70 loci significantly associated with growth stage traits were detected by SSR markers and SNP respectively. In particular, there were stable expression related sites in BLK_29175719-29275719 and BLK_30878620-30978620 on chromosome 13, BLK_48763386-48863386 on chromosome 14 and BLK_10093551-10293551 on chromosome 16. And 11 potential candidate genes related to plant growth and development were predicted in these four regions and four genes (namely,,, and) were directly related to soybean growth period traits.
soybean; growth period; SNP; genome-wide association analysis
10.3724/SP.J.1006.2023.24210
本研究由安徽省重点研发项目(202104a06020029), 国家自然科学基金项目(31871711, 32101704), 四川省科技计划项目(2022SZYZF08),四川省科技计划重点研发项目(2021YFYZ0018), 四川豆类杂粮创新团队春大豆技术研究岗位(SCCXTD-2020-20), 安徽教育厅自然科学基金重点项目(KJ2020A0066)和安徽科技学院项目(2021zrzd13)资助。
This study was supported by the Key Research and Development Projects of Anhui (202104a06020029), the National Natural Science Foundation of China (31871711, 32101704), the Sichuan Science and Technology Program (2022SZYZF08), the Key Research and Development projects of Sichuan (2021YFYZ0018), the Science and Technology Program of Sichuan Province (SCCXTD-2020-20), the Anhui Provincial College Program for Natural Science (KJ2020A0066), and the Key Projects of Anhui Science and Technology (2021zrzd13).
何庆元, E-mail: heqingyuan1@163.com
**同等贡献(Contributed equally to this work)
杨豪, E-mail: 1625763153@qq.com; 向仕华, E-mail: zgxiangshihua@163.com
2022-09-14;
2023-04-17;
2023-04-26.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20230425.1713.008.html
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
猜你喜欢
现代园艺(2017年21期)2018-01-03 06:41:32
中国稻米(2017年2期)2017-04-28 08:00:06
安徽农学通报(2017年1期)2017-02-15 18:37:32
中国稻米(2016年2期)2016-06-29 09:53:29
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
西藏科技(2015年5期)2015-09-26 11:55:25
医学研究杂志(2015年5期)2015-06-10 06:43:26
现代检验医学杂志(2015年5期)2015-02-06 01:42:20
安徽农学通报(2014年6期)2014-04-09 15:28:30
新疆农垦科技(2014年10期)2014-02-28 19:21:18
