
N1-苯基-1,2,3-三氮唑类熊果酸衍生物的设计与合成
2023-08-22 01:18:16魏艳洁赵宪东王欢欢贾宇梦隋丽丽张大军栾天王进
化工与医药工程 2023年4期
魏艳洁,赵宪东,王欢欢,贾宇梦,隋丽丽,张大军,栾天,王进
(沈阳医学院 药学院,辽宁 沈阳 110034)
熊果酸(Ursolic Acid,UA)是一种天然的乌苏烷型五环三萜类化合物。近几年熊果酸的研究在国内外掀起热潮,研究发现其具有广泛的生物学活性,如诱导肿瘤细胞凋亡、抗炎症反应、抗糖尿病、抗心血管疾病、抗病原微生物及抗神经系统疾病等[1]。遗憾的是,熊果酸结构本身存在水溶性差、生物利用度较低等缺点,因此在临床上的应用受到了非常大的限制。为了克服上述缺点,学者们对熊果酸进行结构修饰和改造,从而得到一系列熊果酸衍生物[2-3]。其中,Bilal 等[4]将熊果酸C3 位羟基氧化成酮,得到3-羰基熊果酸(图1 黑色部分),接着将C2 位进行羟醛缩合,引入不同的取代苯甲醛,将所有化合物与3-羰基熊果酸通过噻唑蓝(MTT)法针对人肺癌细胞(A-549),乳腺癌细胞(MCF-7),结肠癌细胞(HCT-116),血癌细胞(THP-1)四种肿瘤细胞以及人正常的上皮细胞(FR-2)进行了抗增殖活性的测试。结果表明,绝大部分化合物针对上述四种肿瘤细胞的IC50值均低于熊果酸,但对FR-2 的IC50值高于熊果酸。其中,化合物1 活性最优,其对A-549,MCF-7,HCT-116,THP-1 的IC50值分别为0.55、0.10、5.5、0.90 μmol/ L。1,2,3-三氮唑因其经典的合成方法被广泛应用于有机合成与药物研发中[5]。报道显示在多种天然产物单体的化学结构中连接1,2,3-三氮唑片段其原有的生物学活性明显提高[6-7]。因此,以熊果酸为结构母体,将其氧化为3-羰基熊果酸,在羰基的邻位引入不同取代的N1-苯基-1,2,3-三氮唑,该片段与化合物1 所拼接的苯基相比,因具有较多的氢键受体而更易与某些靶蛋白相结合,因此通常会具有更为良好的生物活性。本实验通过5 步反应合成4个新化合物,合成工艺成熟,收率理想,1,2,3-三氮唑的合成方法采用经典的Click 环合法,操作简单。本实验合成路线可为相关领域的合成研究提供参考,并为开发三唑类熊果酸衍生物提供借鉴。
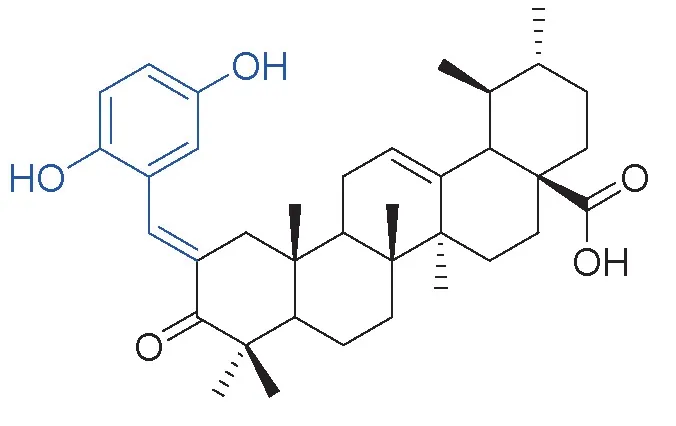
图1 化合物1 的化学结构与生物活性Fig.1 Chemical structure and biological activity of compound 1
1 实验方法
1.1 试剂与仪器
试剂:叠氮化钠、丙炔醇、抗坏血酸钠、熊果酸,阿拉丁试剂、盐酸、亚硝酸钠、氯化钠﹑五水硫酸铜、二氧化锰、三氧化铬、硫酸、氢氧化钾、乙醇、丙酮、异丙醇、石油醚、乙酸乙酯、二氯甲烷、叔丁醇,国药集团化学试剂有限公司。所有试剂均为分析纯。
仪器:DF-II 型集热式磁力加热搅拌器,BSA124S 型电子天平,RE-52 型旋转蒸发器,SHZ-D型循环水式真空泵,WFH-203B 型三用紫外分析仪,Axima CFR plus 型飞行时间质谱仪,Bruker ARX-300核磁共振波谱仪。
1.2 目标化合物的合成
目标化合物的合成路线如图2 所示。
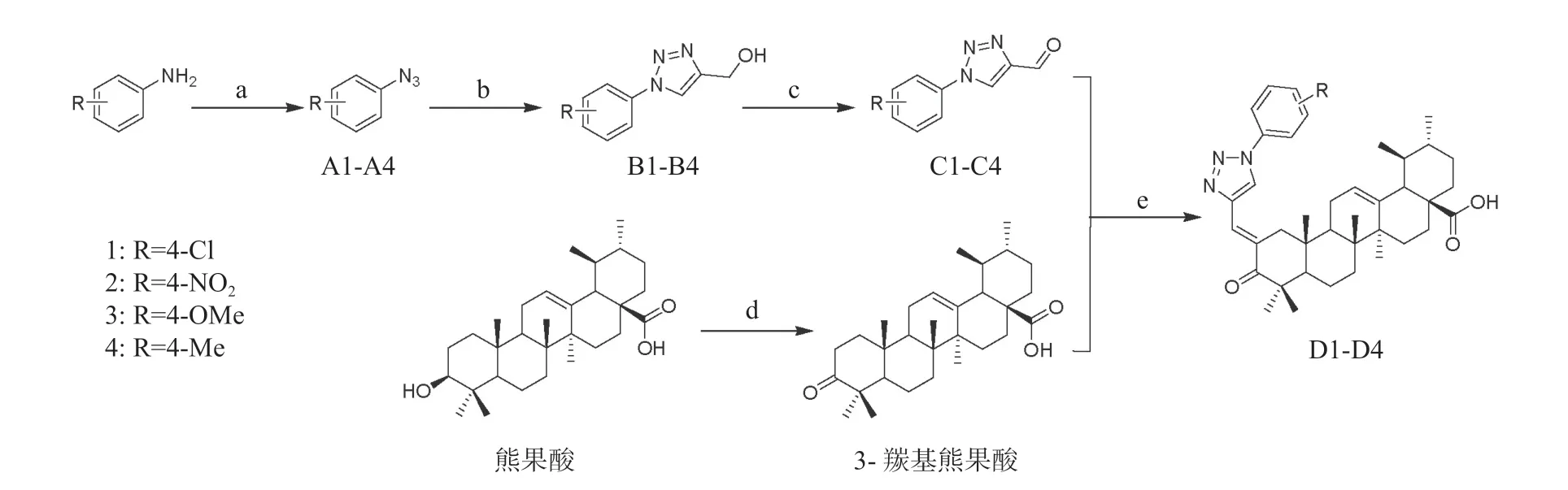
图2 目标化合物的合成路线Fig.2 The synthetic route of the target compounds
1.2.1 中间体A1-A4 的合成
将不同取代的苯胺(10 mmol)溶于10 mL 10%的盐酸中,冰浴下滴加亚硝酸钠溶液(1.035 0 g 的NaNO2溶于25 mL 的水中),滴毕,反应0.5 h 后,滴加叠氮化钠溶液(2.600 0 g 的NaN3溶于50 mL 的水中),3 h 后TLC 监测反应完全(展开剂为石油醚∶乙酸乙酯=15∶1,生成物Rf=0.8);反应液用乙酸乙酯萃取(10 mL× 3),合并有机相,饱和氯化钠溶液洗涤(10 mL × 3)后加入无水硫酸钠干燥,过滤,滤液减压蒸干,得淡黄色油状物,收率95%~ 98%,无需纯化直接进行下一步反应。
1.2.2 中间体B1-B4 的合成
不同取代的中间体A1-A4(9.5 mmol)与0.560 0 g 的丙炔醇溶于35 mL 的叔丁醇中,0.198 0 g的抗坏血酸钠与0.125 0 g 的五水硫酸铜溶于35 mL的水中,合并上述叔丁醇与水溶液,室温下搅拌过夜;反应液用乙酸乙酯萃取(10 mL× 3),合并有机相,饱和氯化钠溶液洗涤(10 mL × 3)后加入无水硫酸钠干燥,过滤,滤液减压蒸干,得白色或淡黄色固体,收率89%~ 92%,无需纯化直接进行下一步反应。
1.2.3 中间体C1-C4 的合成
不同取代的中间体B1-B4(8 mmol)溶于50 mL的丙酮中,加入10.000 0 g 的二氧化锰后回流反应2 h 后TLC 监测反应完全;抽滤,滤液减压蒸干后经硅胶色谱纯化(石油醚-乙酸乙酯=5∶1)得白色固体,收率80%~ 82%。
1.2.4 3-羰基熊果酸的合成
熊果酸(1.0 mmol)溶于60 mL 的丙酮中,冰浴下缓慢滴加琼斯试剂,直至反应液呈现黄红色并在10 min 内不褪色,继续反应1 h,TLC 监测反应完全,滴加1 mL 的异丙醇淬灭琼斯试剂,待反应液呈绿色后抽滤,滤液减压蒸干,加入50 mL 的乙酸乙酯,饱和氯化钠溶液洗涤(10 mL× 3)后加入无水硫酸钠干燥,过滤,滤液减压蒸干,得白色固体0.408 6 g,收率90%。
1.2.5 终产物(D1-D4)的合成
0.090 8 g 的3-羰基熊果酸与中间体C1-C4(物料比为1∶1.1)溶于15 mL 的无水乙醇中,加入0.016 8 g 的氢氧化钾,室温下反应4 h 后TLC 监测反应完全,减压蒸干溶剂,加入30 mL 的水,用稀盐酸调节pH 至酸性,用乙酸乙酯萃取(10 mL× 3),合并有机相,饱和氯化钠溶液洗涤(10 mL× 3)后加入无水硫酸钠干燥,过滤,滤液减压蒸干后经硅胶色谱纯化(石油醚-乙酸乙酯=5∶1)得白色固体收率76%~ 79%。
2 结果与讨论
2.1 目标化合物的合成
本实验以不同取代的苯胺为起始物料,第一步为“一锅法”,首先在酸性条件下被亚硝酸钠还原为重氮苯,再被叠氮化钠叠氮化生成不同的叠氮苯,本反应应严格控制叠氮化钠溶液的滴速,防止过度放热。第二步以中间体A 为原料,通过Click 反应制得中间体B,将二价铜离子被抗坏血酸钠还原为一价铜离子后催化环合反应;本反应探索了反应溶剂对反应的影响,发现使用叔丁醇﹑四氢呋喃这类与水互溶的有机试剂或二氯甲烷等与水分层的有机试剂均可发生该反应,本反应副产物少,目标产物易于纯化。第三步使用二氧化锰作为弱氧化试剂将不同的醇(中间体B1-B4)氧化为醛,本反应在后处理中应在抽滤滤纸上加适宜量的硅藻土,防止二氧化锰颗粒过细透过滤纸。最后将不同取代的醛(中间体C1-C4)在碱性条件下与3-羰基熊果酸通过克莱森-施密特缩合反应得终产物,因3-羰基熊果酸与终产物极性较接近,与终产物较难通过色谱法分离,因此反应过程中使中间体C 稍过量,过量的中间体C 可通过硅胶柱层析与终产物进行分离,产品易于纯化。
2.2 目标化合物的结构表征
目标化合物的质谱及核磁共振波谱数据如表1及图3~ 5 所示,以D1 为例,在核磁氢谱数据中,化合物的化学位移8.05 的单重峰为1,2,3-三氮唑C5 位上的氢原子峰,当苯环上引入硝基等强吸电子效应基团时,其化学位移相对较大(如D2 为8.18)。结合耦合常数可以确定,化学位移7.74 的双重峰为与三唑相连苯环邻位上的两个氢原子峰,7.56 的双重峰为与三唑相连苯环间位上的两个氢原子峰,化学位移7.50 的单重峰为与1,2,3-三氮唑C4 位相连碳原子上的氢原子峰,化学位移5.35 的单重峰为熊果酸母体C12 位双键上的氢原子峰,其余均为熊果酸环上或角甲基的氢原子峰;结合核磁共振碳谱的碳原子总数以及质谱的分子量测试结果表明目标化合物的结构均正确。
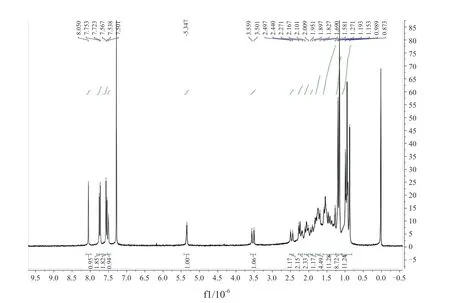
图3 化合物D1 的核磁共振氢谱图Fig.3 1H-NMR spectra of compound D1
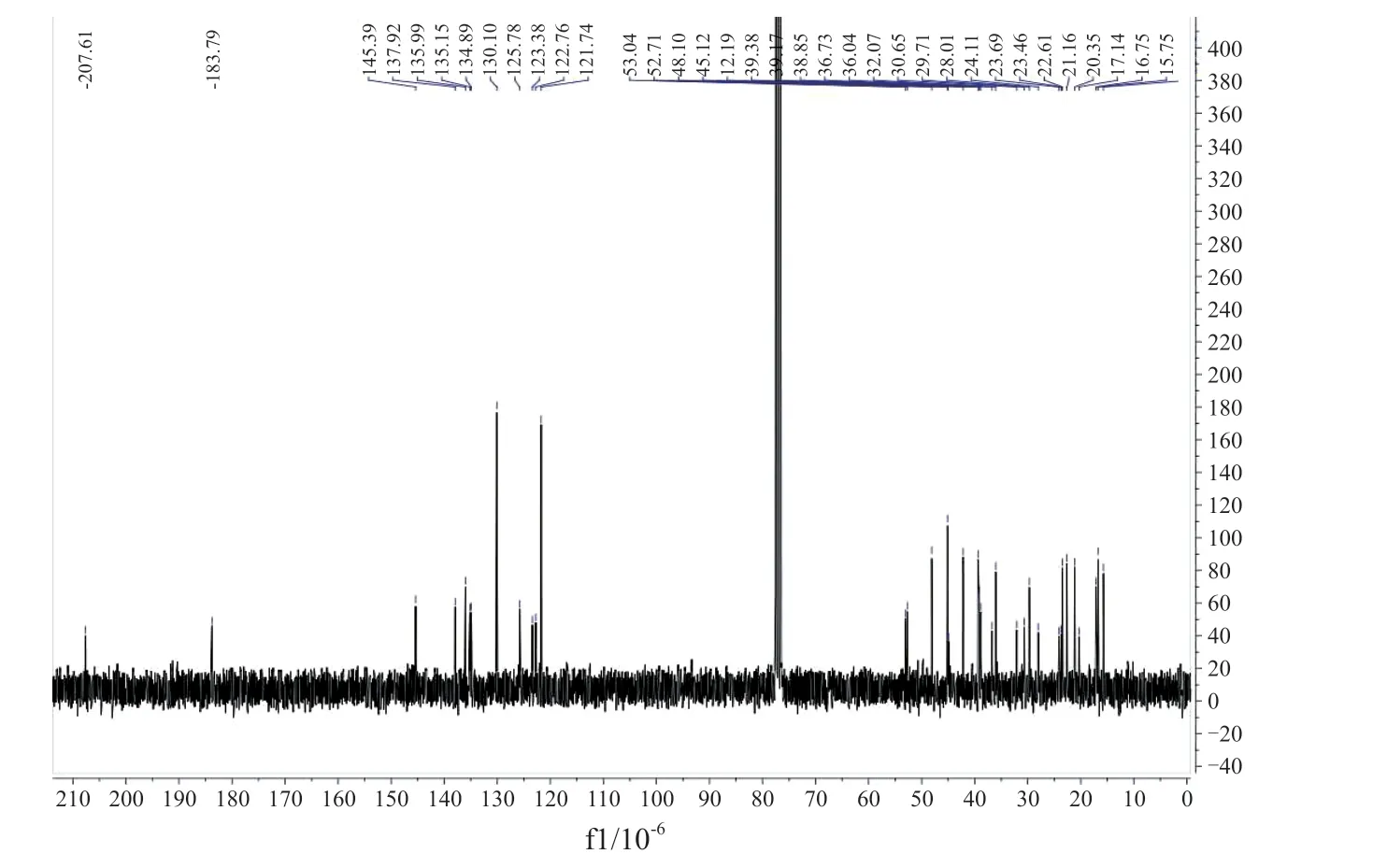
图4 化合物D1 的核磁共振碳谱图Fig.4 13C-NMR spectra of compound D1
图5 化合物D1 的质谱图Fig.5 Mass spectra of compound D1
3 小结
经5 步反应制得4 个未见文献报道的新型熊果酸衍生物,总收率46%~ 53%。本实验的研究内容可为三唑类熊果酸衍生物的合成提供一定的参考。在后续的研究中可通过对该类化合物的生物学活性进行探究,为新药的开发与研制提供有价值的借鉴。
猜你喜欢
江南诗(2023年6期)2023-12-08 05:17:24
金沙江文艺(2022年1期)2022-02-04 10:15:16
陶瓷学报(2021年5期)2021-11-22 06:35:34
化学教与学(2021年12期)2021-02-18 01:16:58
中学生数理化(高中版.高考理化)(2020年1期)2020-11-24 21:02:17
商品与质量(2019年32期)2019-11-29 05:56:00
火工品(2018年1期)2018-05-03 02:27:56
中国资源综合利用(2017年3期)2018-01-22 02:45:40
合成化学(2015年9期)2016-01-17 08:57:14
应用化工(2014年1期)2014-08-16 13:34:08
