
干旱胁迫下忍冬全基因组DNA甲基化和转录组分析
2023-08-17 12:59许小涵唐志强刘振华张永清蒲高斌
中草药 2023年16期
许小涵,唐志强,刘 谦, 2,李 佳, 2,刘振华, 2,张永清, 2,蒲高斌, 2*
• 药材与资源 •
干旱胁迫下忍冬全基因组DNA甲基化和转录组分析
许小涵1,唐志强1,刘 谦1, 2,李 佳1, 2,刘振华1, 2,张永清1, 2,蒲高斌1, 2*
1. 山东中医药大学,山东 济南 250355 2. 山东省中药质量控制与全产业链建设协同创新中心,山东 济南 250355
分析干旱胁迫下忍冬全基因组DNA甲基化水平变化、模式以及与基因表达的关系,为进一步探究忍冬响应干旱胁迫的分子机制奠定基础。采用全基因组重亚硫酸盐甲基化测序法(whole-genome bisulfite sequencing,WGBS),测定干旱胁迫下忍冬全基因组DNA甲基化水平,并联合转录组测序结果对差异甲基化基因相关的差异表达基因进行分析。干旱胁迫下忍冬整体甲基化水平普遍上升;其中CG类型甲基化水平明显高于CHG或CHH甲基化类型;对不同干旱胁迫处理下的忍冬进行差异甲基化区域(differentially methylated regions,DMR)分析,发现1935、2158和1750个差异甲基化相关基因,基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)富集分析表明,显著富集的通路主要包括亚油酸代谢,氮代谢通路和次生代谢物的生物合成通路等。通过转录组测序技术,发现差异甲基化基因相关的差异表达基因主要富集于次生代谢产物合成相关通路,同时筛选出4个在干旱胁迫下基因表达量上升显著的忍冬MYB(MYB,LjMYB)转录因子。干旱胁迫下忍冬甲基化水平上升,推测DNA甲基化调控基因表达是干旱影响金银花有效成分生物合成的作用机制之一。
忍冬;干旱胁迫;DNA甲基化;全基因组重亚硫酸盐甲基化测序;转录组测序
金银花为忍冬科忍冬属植物忍冬Thunb.的干燥花蕾或带初开的花,具有清热解毒、疏散风热之功效[1],为常用大宗中药材之一。目前,金银花商品药材主要来自人工栽培,其中山东省的种植面积近6.667万公顷,为主要道地产区。现代生物学认为,道地药材的本质是药用植物所拥有的基因型受环境因子诱导后表达的产物[2]。受环境影响,金银花药材质量差异较大。如金银花“九丰一号”由山东曲阜引种至广西武鸣后,在相同采摘时期和干燥条件下,其木犀草苷含量下降了57.4%[3]。另有研究表明,水分[4]、光照[5]、温度[6-7]等环境因子对金银花次生代谢产物的生物合成均有显著影响。然而,环境因子究竟是如何通过基因型修饰而发生作用的,特别是环境因子影响金银花有效成分生物合成的作用机制,至今缺少直接的实验研究的揭示和证明。
表观遗传学研究为揭示环境与药材品质的关系提供了契机。表观遗传学是以不改变基因DNA序列编码的方式来改变遗传基因表达的一种遗传方式,主要涉及DNA甲基化作用的改变、RNA沉默、染色质组蛋白的修饰作用、基因印记[8]。DNA甲基化是表观遗传学的重要研究内容之一,与基因表达、基因组防御、细胞分化、染色质失活和基因组印记的调节有关[9]。研究发现红光和远红光照射沉香后,其葫芦素含量发生改变,通过全基因组重亚硫酸氢盐测序发现红光和远红光条件下甲基化水平变化较大,红光可能通过改变沉香中CHH和CHG的甲基化来调控基因的表达[10]。谢宜杰等[11]通过MASP技术测定广藿香DNA甲基化水平,能够在不同来源的样品中鉴别出石牌产地的广藿香。Li等[12]采用HPLC法测定红豆杉不同传代培养细胞中全基因甲基化水平,发现DNA总体甲基化水平高低与紫杉醇的量呈显著的负相关关系,使用5-azaC处理后细胞中紫杉醇积累量显著增加。Zha等[13]研究发现忍冬Thunb. 和其自然诱变产生的变种红白忍冬Thunb. var.(Wats.) Bak.绿原酸含量在两者之间差异显著,通过测定DNA甲基化水平发现红白忍冬苯丙氨酸裂解酶2基因侧翼区域的启动子−109~−279 bp处DNA甲基化程度要高于忍冬,且二者间CG位点的DNA甲基化程度显著不同。
可见,探明忍冬植株对干旱胁迫的应答机制,对于稳定金银花药材质量、指导金银花生产、发展金银花生态种植具有重要意义。然而,相关研究尚未见报道。本研究利用重亚硫酸盐测序法测定在甘露醇模拟干旱胁迫环境下忍冬叶片的DNA 甲基化水平,并分析其甲基化模式,阐述干旱胁迫对于忍冬全基因组DNA甲基化水平变化的影响,同时结合转录组数据分析干旱胁迫下忍冬DNA甲基化差异区域与表达水平差异基因之间的调控关系。研究结果将为今后忍冬新型植物分子抗旱育种及干旱胁迫下忍冬表观遗传变化的分子机制奠定基础。
1 材料与仪器
1.1 材料
样品采自经山东中医药大学药草园,由山东中医药大学蒲高斌教授鉴定为忍冬科忍冬属植物忍冬Thunb.
1.2 仪器
CFX96 Touch Real-Time PCR仪(Bid-Rad 公司);5424D型高速离心机(Eppendorf 公司);甘露醇购自上海源叶生物科技有限公司;Hoagland’s 营养液购自青岛海博生物技术有限公司;多糖多酚植物RNA提取试剂盒购自南京诺唯赞生物科技股份有限公司;SYBR Preminx EX TaqTM(ROX)、RT 反转录试剂盒购自TaKaRa公司。
2 方法
2.1 样品处理
全基因组甲基化及转录组测定材料为3年生“华金2号”忍冬扦插苗。将园土与基质按1∶1比例混合,基质含草炭土、木质泥炭、优等椰壳粉、蛭石、高岭石及有机物质和生长所需的营养物质,装入内径为38 cm的花盆中。选择生长情况良好、大小近似的忍冬苗移入花盆内,每盆1株,植物在温室条件中生长,进行常规水分管理。向每盆扦插苗中浇灌2 L 0.2 mol/L的甘露醇用来进行模拟干旱处理,在处理的0(CK)、3(DS-3d)、6(DS-6d)、9 d(DS-9d)分别取植株基本一致位置的幼叶,并立即用液氮冷冻,放入−80 ℃冰箱中备用。
MYB转录因子表达模式分析采用“华金2号”忍冬幼苗。培养条件为光照培养16 h、暗培养8 h,温度为25 ℃。待长至第6片叶时,将材料分成2组,一组设置为对照,一组在培养液中加入甘露醇使其终浓度为0.2 mol/L,分别选取对照及干旱胁迫处理1、3、8、12、24 h植物材料的叶片,液氮冷冻后放入−80 ℃冰箱中保存备用。
2.2 DNA的提取、质检、建库及测序
利用植物DNA提取试剂盒提取不同干旱胁迫下样品DNA,DNA样品经广州基迪奥生物科技有限公司质检合格后构建亚硫酸盐(BS-seq)文库构建,测序步骤为(1)超声处理DNA破碎为100~300 bp的片段。(2)DNA片段末端修复,并在3’端加上A碱基,然后将甲基化测序接头连接到基因组片段上。(3)采用ZYMO EZ DNA Methylation- Gold kit进行Bisulfite处理,Bisulfite处理会使未甲基化的胞嘧啶转化为尿嘧啶。(4)脱盐处理后切胶回收并进行文库片段大小选择,PCR扩增后重复大小选择;构建完成后,检测质量。最后使用Illumina Hi-SeqTM2500平台测序。
2.3 测序数据过滤及比对分析
测序后得到原始数据,从原始数据去除含N比例大于10%、低质量的reads后得到高质量干净读数(clean data)。使用BSMAP(version:2.90)软件将clean data比对到参考基因组序列上,选取唯一比对到的序列进行下一步分析。参考基因组选用忍冬基因组,源自国家基因组数据中心(National Genomics Data Center,https://ngdc.cncb.ac.cn/ gwh/),登录号为GWHAAZE00000000[14]。
2.4 甲基化水平及差异甲基化区域分析
对不同处理组之间进行甲基化分析,主要是统计不同处理下样品全基因组的甲基化水平和特定元件甲基化水平。使用methylkit软件(version:1.7.10)进行DNA差异甲基化区域(differentially methylated regions,DMR)分析,采用200 bp窗口在全基因组扫描,统计各个窗口内特殊位点的平均DNA甲基化率,比较差异。
大学英语教学有其自身的特点:从教学内容来看,大学英语教学内容具有前沿性和实践性;从教学管理来看,大学的管理模式与中学有很大的差异,学生的自由支配时间很多;从大学生学习过程的特点来看,学生可以根据自身未来发展的需要,制定学习计划和目标,在学习的过程中不断钻研,充分发挥学习的自主性,以弥补课堂教学中欠缺的在认知风格、学习策略、学习动机以及能力方面的差异,以实现自我职业发展的需求。
2.5 差异甲基化区域相关基因功能富集分析
得到全基因组DNA甲基化数据后,可以通过对DMR相关基因进行基因本体(gene ontology,GO)和基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)通路富集分析,了解显著富集的生物学功能,从而推断出干旱胁迫对忍冬基因表达的影响[15]。
2.6 转录组测序
转录组数据为本课题组前期实验所得。利用植物RNA提取试剂盒提取样品RNA,RNA样本检测合格后进行普通转录组文库构建,文库检测合格后,利用Illumina NovaSeq6000进行测序。对测序结果进行过滤,去除掉低质量读数后得到高质量的clean data,将得到的clean data与参考基因组进行比对,参考基因组同甲基化测序参考基因组。利用featureCounts(1.5.0-p3)计算映射到每个基因的读数,然后根据基因的长度计算每个基因的FPKM,并计算映射到该基因的读数,以此来定量表示基因表达水平。基于基因表达定量分析得到数据,采用edgeR进行基因差异表达显著性分析,根据log2|(fold change)|>1且值<0.05,得到不同干旱胁迫处理下与对照组相比的差异表达基因(differentially expressed gene,DEG)。
2.7 全基因组DNA甲基化联合转录组分析
根据不同处理组全基因组DNA甲基化数据中meth.diff值将差异甲基化基因分为甲基化水平上升组(A1)和甲基化水平下降组(A2)。根据不同处理组转录组数据中基因的FPKM值和log2FC值将差异表达基因分为表达水平上升组(B1)和表达水平下降组(B2),综合分析数据。
2.8 干旱胁迫下忍冬MYB(L. japonica MYB,LjMYB)转录因子表达模式分析
从“2.7”项结果中,筛选出甲基化水平下降同时转录组水平上升的转录因子,利用qRT-PCR验证干旱胁迫下MYB转录因子的表达情况,基因序列在网站[Home-Genome Warehouse(www.cncb.ac.cn)]中查找,利用Primer 3设计Real-Time PCR引物,内参基因为金银花Lj18S,所用引物铂尚生物技术有限公司负责合成。将对照以及干旱胁迫处理1、3、8、12、24 h叶片加液氮研磨成粉末,利用多糖多酚植物RNA提取试剂盒(南京诺唯赞)提取RNA,采用反转录试剂盒(Takara)将RNA反转录为cDNA。以反转录后的cDNA为模板,利用SYBR试剂盒(Takara)进行qRT-PCR。反应体系25 μL:TB Green Premix Ex TaqⅡ(2×)12.5 μL,正向引物1 μL,反向引物1μL,cDNA模板2 μL,灭菌水8.5 μL。反应程序为:95 ℃预变性3 min;95 ℃变性10 s,54 ℃退火30 s、72 ℃延伸1 min,39个循环,65~95 ℃过程中每隔0.5 ℃作溶解曲线,设3个重复。采用2−ΔΔCt法计算基因的相关表达水平。
3 结果与分析
3.1 亚硫酸氢盐测序总结
采用全基因组重亚硫酸氢盐甲基化测序测定忍冬在不同干旱胁迫处理下DNA甲基化水平,利用Illumina Hi-Seq 2500测序平台测序,测序数据处理及与参考基因组的比对结果见表1。在不同处理时间下,过滤测序得到的原始数据,分别得到304 537 910、273 790 354、185 030 316、207 805 832个clean reads。使用BSMAP比对数据与基因组序列,比对率分别为83.60%、83.31%、83.66%、84.39%,表明该方法和结果具有较高的可靠性和准确性。此外,基因组的有效覆盖率在70%以上,不同甲基化类型之间甲基化水平存在明显差异如图1所示。
表1 重亚硫酸氢盐测序结果
Table 1 Results of bisulfite sequencing
样品名总序列读数比对序列读数比对率/%亚硫酸盐处理转化率/%比对深度覆盖率/% CK304 537 910254 578 51183.6099.4942.2573.50 DS-3d273 790 354228 083 36583.3199.5037.8572.49 DS-6d185 030 316154 798 26583.6699.5825.6971.07 DS-9d207 805 832175 362 37684.3999.5229.10 72.67

图1 不同序列环境甲基化占比
3.2 忍冬DNA甲基化的全基因组模式
3.2.1 忍冬全基因组DNA甲基化分析 全基因组平均甲基化水平由有效覆盖C碱基的甲基化水平之和与有效覆盖碱基总数之比决定,甲基化胞嘧啶的百分比根据局部序列环境和外部处理而变化。在CK、DS-3d、DS-6d、DS-9d 4种环境下,忍冬全基因组mC水平分别为23.60%、26.97%、28.37%、25.06%。与CK相比在干旱处理3、6 d时,全基因组甲基化水平会随着干旱时间的延长不断上升。9 d时,虽然甲基化水平与3、6 d相比有所下降,但仍然高于CK组。
CK组处理的基因组中mCG、mCHG、mCHH的甲基化水平分别为79.84%、50.95%、11.42%(表2),这反映了忍冬基因组中甲基化水平的百分比。同时,通过分析mCG、mCHG和mCHH序列相对于总mC位点出现的比例,可以反映出mC位点在3种序列环境中的分布。如图1所示,甲基化胞嘧啶最常出现在mCHH位点(60.01%),在mCG、mCHG序列出现频率较低,分别为23.64%和16.31%。
表2 甲基化水平统计
Table 2 Statistical table of methylation level
样品名C/%CG/%CHG/% CHH/% CK23.6079.8450.9511.42 DS-3d26.9780.3852.4615.46 DS-6d28.3781.3954.2016.56 DS-9d25.0680.5351.7512.79
对于每一区域来说,在3种不同序列环境中CG位点的平均甲基化水最高,而CHH序列环境则呈现出最低的甲基化水平。具体来说,在Up2k区域发生甲基化水平较高即启动子区域甲基化水平较高,其次是内含子和外显子区域,在CG序列环境下,内含子区域CG平均甲基化水平最高,其次为外显子区域与启动子区域。在CHG序列环境下,启动子区域CHG平均甲基化水平最高,远高于外显子和内含子区域。此外,CHH序列环境下各基因组区域的DNA甲基化水平模式与CHG序列环境基本类似。干旱胁迫后,各基因组区域DNA甲基化水平模式基本未变,但在甲基化率方面有所变化,启动子区域中,CHH类型甲基化率有所上升;外显子区域甲基化率基本未变,内含子区域各甲基化类型甲基化水平均有小幅上升(图2)。
3.3 胞嘧啶DNA甲基化序列偏好性分析
在真核生物中,全基因组范围内甲基化位点附近碱基的序列特征,对了解甲基化发生的序列偏好有重要作用。为了揭示序列环境和忍冬甲基化偏好之间是否存在关系,使用Logo Plots的工具对CHG碱基进行统计,得到全基因组中CHG与mCHG碱基的附近9 bp的碱基构成,通过CHG到mCHG的比较,探索甲基化位点或其附近区域的序列信息。在所有甲基化的mC位点中,mC位点周围的序列偏好随着CG背景和非CG背景而变化(图3),在对称CG环境中,mC经常出现在TCGA序列中,而mCHG背景下mC位点的甲基化最经常出现在CAG序列中,而mCHH范围内的甲基化最常发生在CAA序列中。因此,推断mC位点的大多数甲基化发生在CAN(N代表A、T、C、G)。
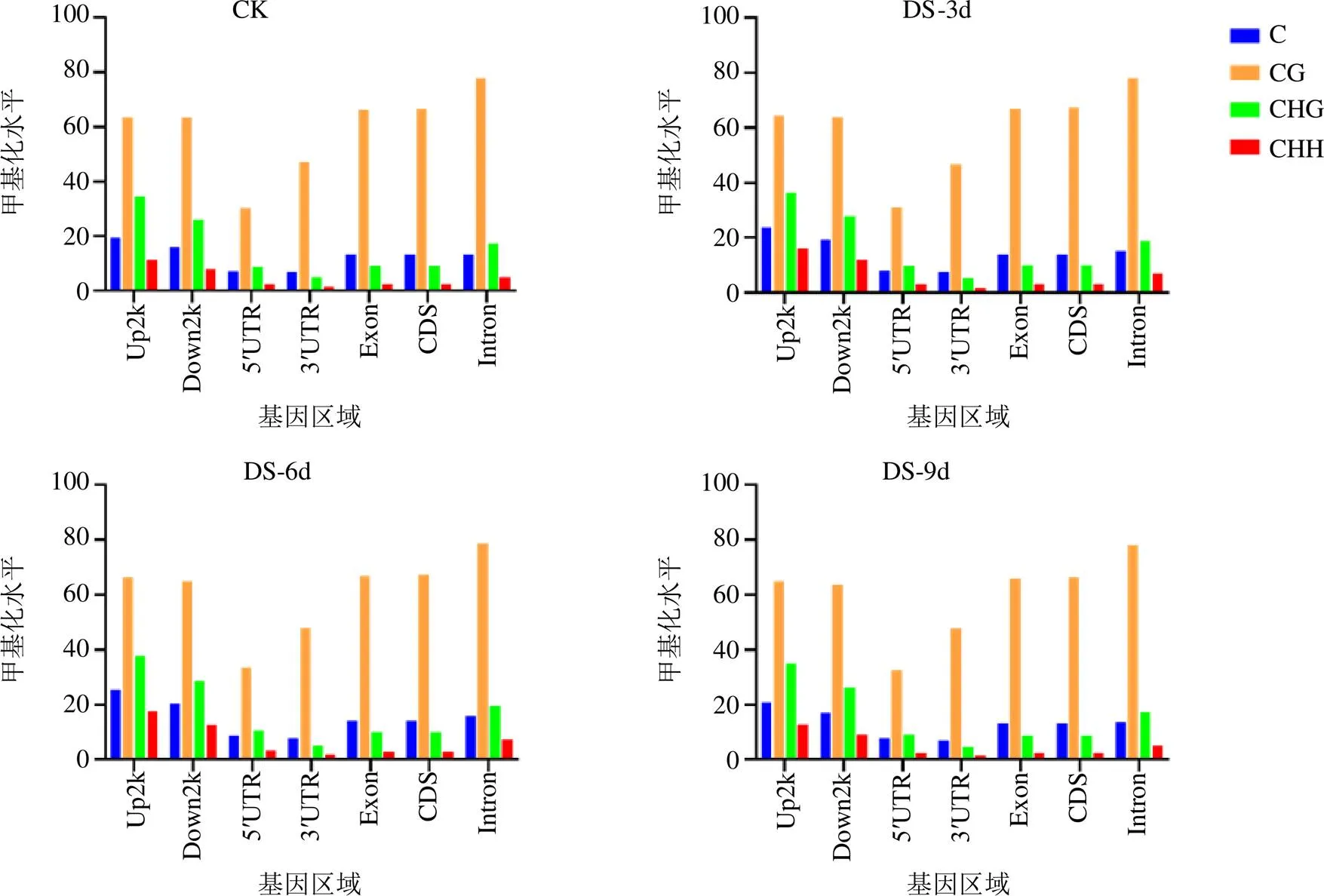
Up2k为基因上游2 kb,Down2k为基因下游2 kb,5’UTR为5’非翻译区,3’UTR为3’非翻译区,Exon为外显子,CDS为编码序列,Intron为内含子
3.4 忍冬基因组的DMR及相关基因富集分析
3.4.1 DNA甲基化的DMR分析 将不同干旱处理组与对照组进行比较,分别检测出5926、6978和5172个DMR(图4)。对其进行分析发现,在DS-3d组中,有3953个DMR甲基化水平下降,有1973个DMR甲基化水平上升。在DS-6d组中,有2068个DMR甲基化水平下降,有4910个DMR甲基化水平上升。在DS-9d组中,有3224个DMR甲基化水平下降,有1948个DMR甲基化水平上升。与对照组相比,处理组甲基化水平下降DMR数量均大于甲基化水平上升DMR数量。
3.4.2 DMR相关基因富集分析 将前期得到的DMR相关基因进行GO和KEGG富集分析,挖掘出与忍冬在干旱胁迫条件下化学成分变化相关的生物学过程和pathway调控通路,进而探讨差异甲基化基因在忍冬响应干旱胁迫中的调控作用。
GO富集分析结果显示在DS-3d、DS-6d、DS-9d处理下,分别有478、557和464个显著富集的GO条目(<0.05),分别来自于193、2158和1750个DMR相关基因,称为DMG(差异性甲基化相关基因)。将3组比较组的差异甲基化基因富集到GO的3类中,其二级注释如图5所示,在细胞组成项下,DMG主要富集到细胞,细胞组分和细胞器。在生物过程项下,DMG主要富集到细胞过程,代谢过程和单一生物过程。在分子功能项下,DMG主要富集到整合,催化反应和运输反应,表明甲基化介导忍冬中这些类型的途径。
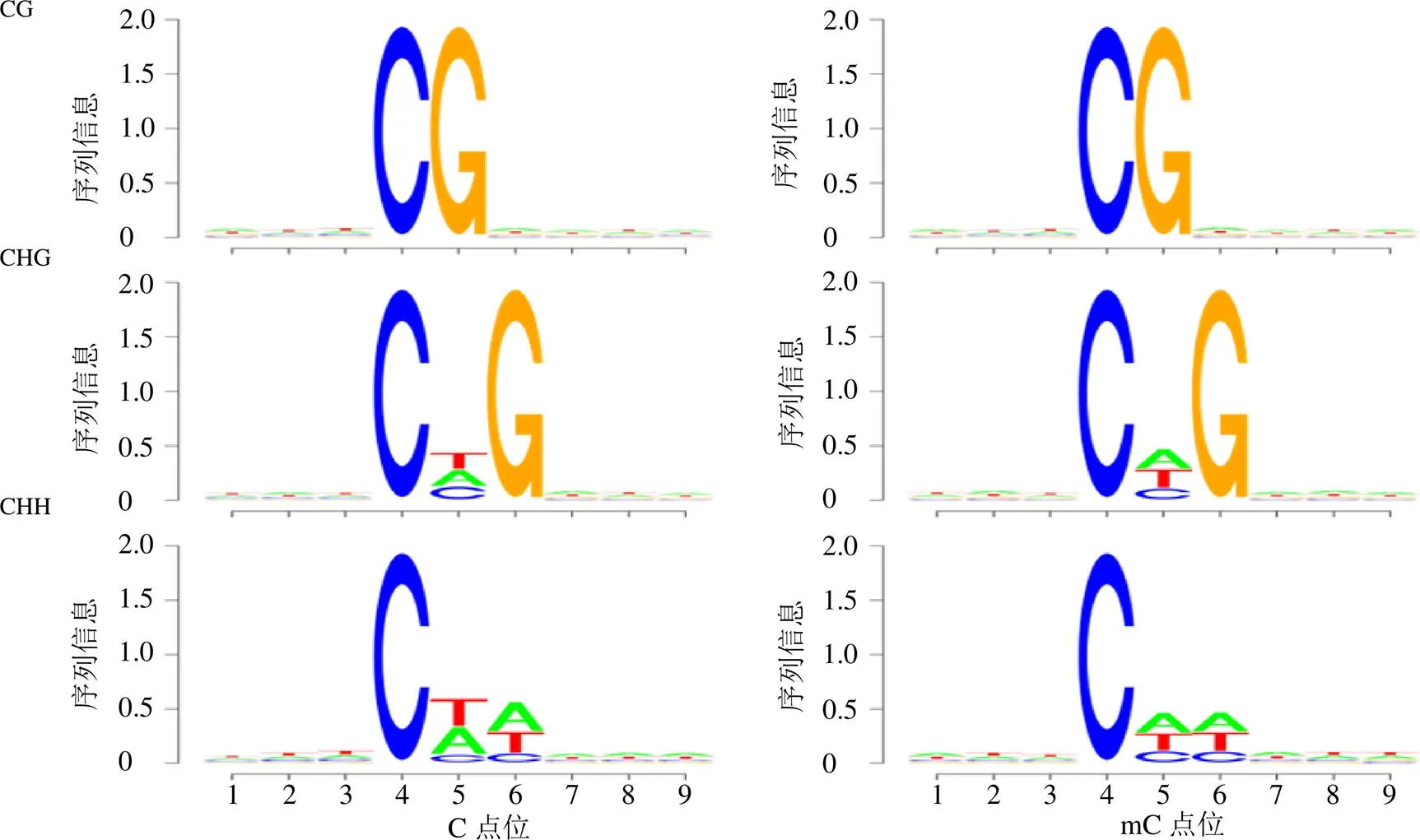
X轴表示甲基化胞嘧啶位于第4位的碱基位置,Y轴表示碱基的熵
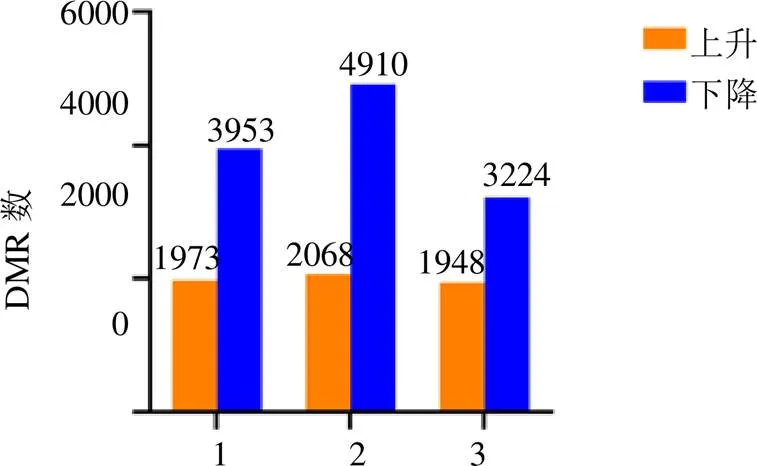
1-DS-3d组与CK相比 2-DS-6d组与CK相比 3-DS-9d组与CK相比
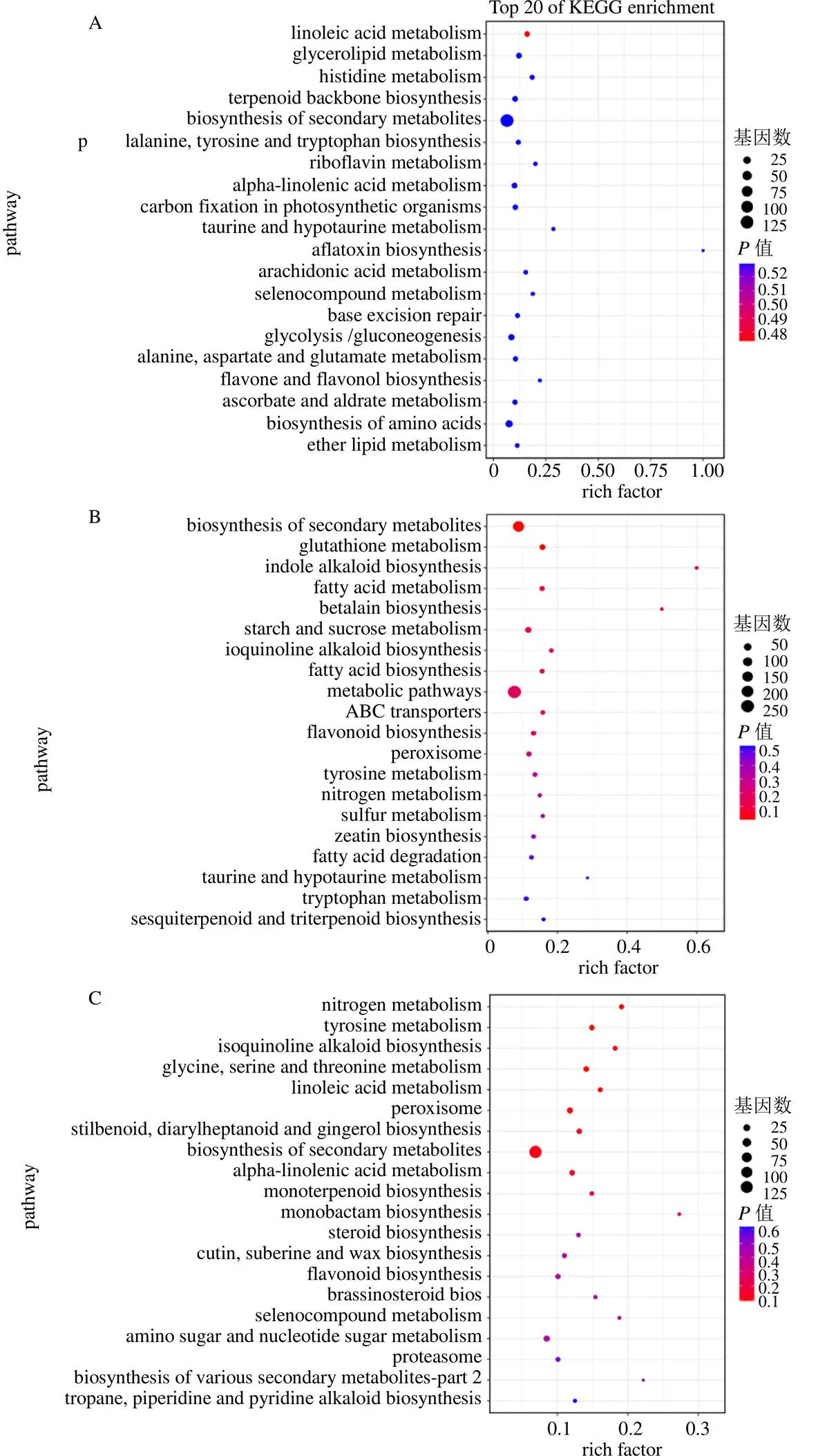
KEGG富集分析显示(图6),在DS-3d、DS-6d、DS-9d处理下分别有7、15和12条通路显著富集(<0.05)。DS-3d组与CK相比,中显著富集的pathway包含亚油酸代谢、甘油酯代谢、组氨酸代谢、萜类生物合成、苯丙氨酸、酪氨酸和色氨酸生物合成以及次生代谢物的生物合成等7条通路,涉及到387个相关基因。DS-6d组与CK相比,显著富集的pathway包括次生代谢物的生物合成、谷胱甘肽代谢、脂肪酸代谢、异喹啉生物碱生物合成、淀粉与蔗糖的代谢、脂肪酸生物合成以及ABC转运黄酮类生物合成等15条通路,涉及468个相关基因。DS-9d组与CK相比,显著富集的途径包括氮代谢、酪氨酸代谢、异喹啉生物碱生物合成、亚油酸代谢、过氧化物酶、二苯乙烯、二芳基庚烷和姜酚的生物合成和次生代谢物的生物合成等12条通路,涉及393个相关基因,这些途径可能与忍冬对干旱的响应密切相关。
3.5 转录组数据分析
对照组及不同干旱胁迫处理组测序结果经过滤后,分别得到44 402 319、44 655 626、43 873 937、42 504 058个clean reads,与参考基因组的比对率分别为93.28%、91.98%、92.36%、92.43%。通过与对照组相比,在DS-3d、DS-6d、DS-9d分别鉴定出8531、4171和6712个差异表达基因。其中在DS-3d时,有4126个基因表达上升,4405个基因表达下降。在DS-6d时,有2849个基因表达上升,1322个基因表达下降。在DS-9d时,有3727个基因表达上升,2985个基因表达下降。
3.6 全基因组DNA甲基化与转录组联合分析
为了探讨干旱胁迫期间观察到的mC甲基化变化是否会改变基因表达,综合分析差异甲基化基因和差异表达基因的数据。在DS-3d时有82个高表达基因其甲基化水平升高以及264个高表达基因其甲基化水平降低;在DS-6d时有56个高表达基因其甲基化水平升高以及341个高表达基因其甲基化水平降低(图7);在DS-9d时有96个高表达基因其甲基化水平升高以及237个高表达基因其甲基化水平降低;前期KEGG富集分析显示,这些基因广泛参与次级代谢产物的生物合成、苯丙烷代谢、类黄酮生物合成、醌和其他萜醌生物合成、异喹啉生物碱生物合成、MAPK信号通路-植物、激素信号转导、倍半萜类和三萜类生物合成、萜类骨架生物合成以及二萜类生物合成等,由此推测干旱胁迫下忍冬可通过体内DNA甲基化水平的改变调控次生代谢产物合成途径中相关基因的表达。

A-DS-3d组与CK相比 B-DS-6d组与CK相比 C-DS-9d组与CK相比;红色代表生物过程类,绿色代表细胞组成类,蓝色代表分子功能类
A-DS-3d组与CK相比 B-DS-6d组与CK相比 C-DS-9d组与CK相比
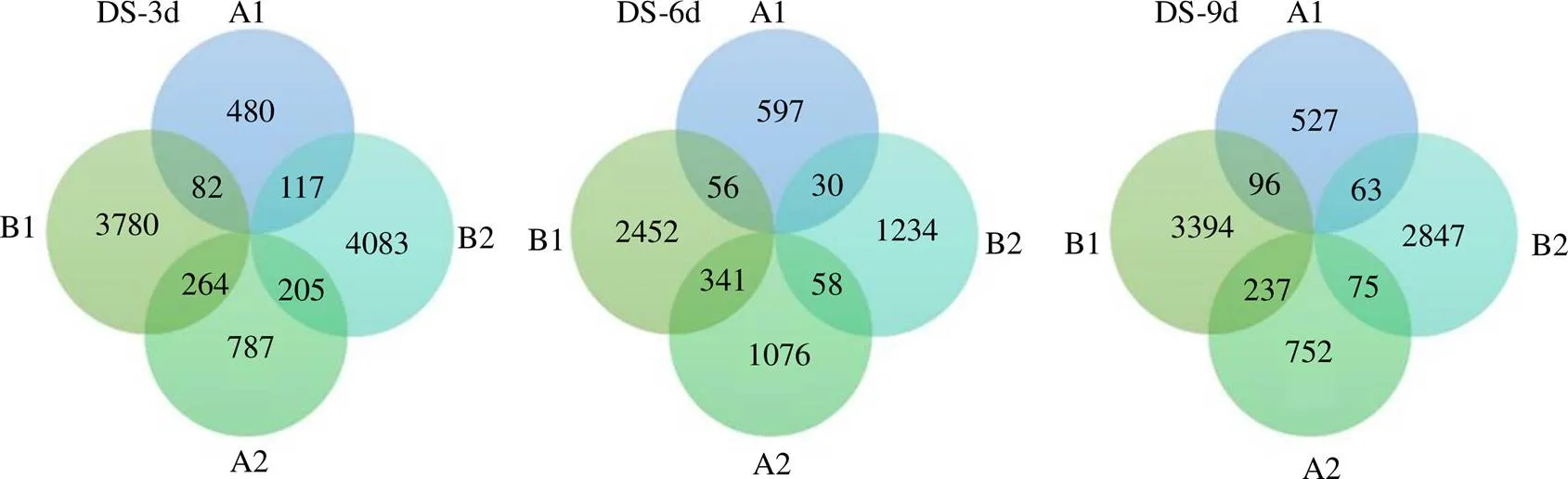
A1-甲基化水平上升DMR A2-甲基化水平下降DMR B1-表达水平上升DEG B2-表达水平下降DEG
3.7 干旱胁迫下LjMYB转录因子表达模式分析
在DS-3d、DS-6d、DS-9d 3个处理组转录组水平上升且甲基化水平下降的基因中共筛选出10个MYB转录因子,分别命名为~。利用qRT-PCR对10个转录因子在对照以及0.2 mol/L甘露醇处理不同时间下的表达模式进行分析。
qRT-PCR结果如图8所示,干旱胁迫下转录因子的表达各不相同,基因表达水平整体呈上升趋势。其中和基因表达水平在胁迫处理3 h时差异显著,分别升高13.19倍和7.73倍,和在胁迫处理24 h时差异显著,分别升高9.13倍和7.64倍。在干旱胁迫8 h时,基因表达水平达到最高,而、在胁迫处理24 h表达量最高。、、在不同胁迫时间下表达量无显著差异。结果表明在干旱胁迫处理不同时间下转录因子的变化有所不同,是一个动态的变化,推测在干旱胁迫下植物可通过改变MYB转录因子的甲基化水平来调控体内成分变化以应对胁迫。
4 讨论
全基因组重亚硫酸盐甲基化测序法(whole- genome bisulfite sequencing,WGBS)技术作为基因组甲基化测定的一种方法,因具有精确度高、结果可靠及单碱基分辨率等优点成为DNA甲基化检测的“金标准”[16]。目前在已经进行基因组DNA甲基化分析的34种植物中,mCG在全基因组范围内具有最高水平的DNA甲基化,拟南芥mCG在已测物种中最低,为30.5%,甜菜 mCG最高,为92.5%。CHG位点甲基化水平变化范围从9.3%的盐水水芹到81.2%的甜菜。葡萄中mCHH含量最低,仅为1.1%,甜菜中mCHH含量最高,为18.8%[17-18]。本研究利用WGBS技术测定忍冬在不同干旱胁迫条件下DNA甲基化水平,CG位点甲基化水平从79.84%~81.39%,CHG位点甲基化水平从50.95%~54.20%,CHH位点甲基化水平从11.42%~16.56%。与其他物种相比,忍冬DNA甲基化水平在变化范围之内,且忍冬基因组各序列环境具有较高的甲基化水平,甲基化水平随着干旱胁迫条件的改变而变化。
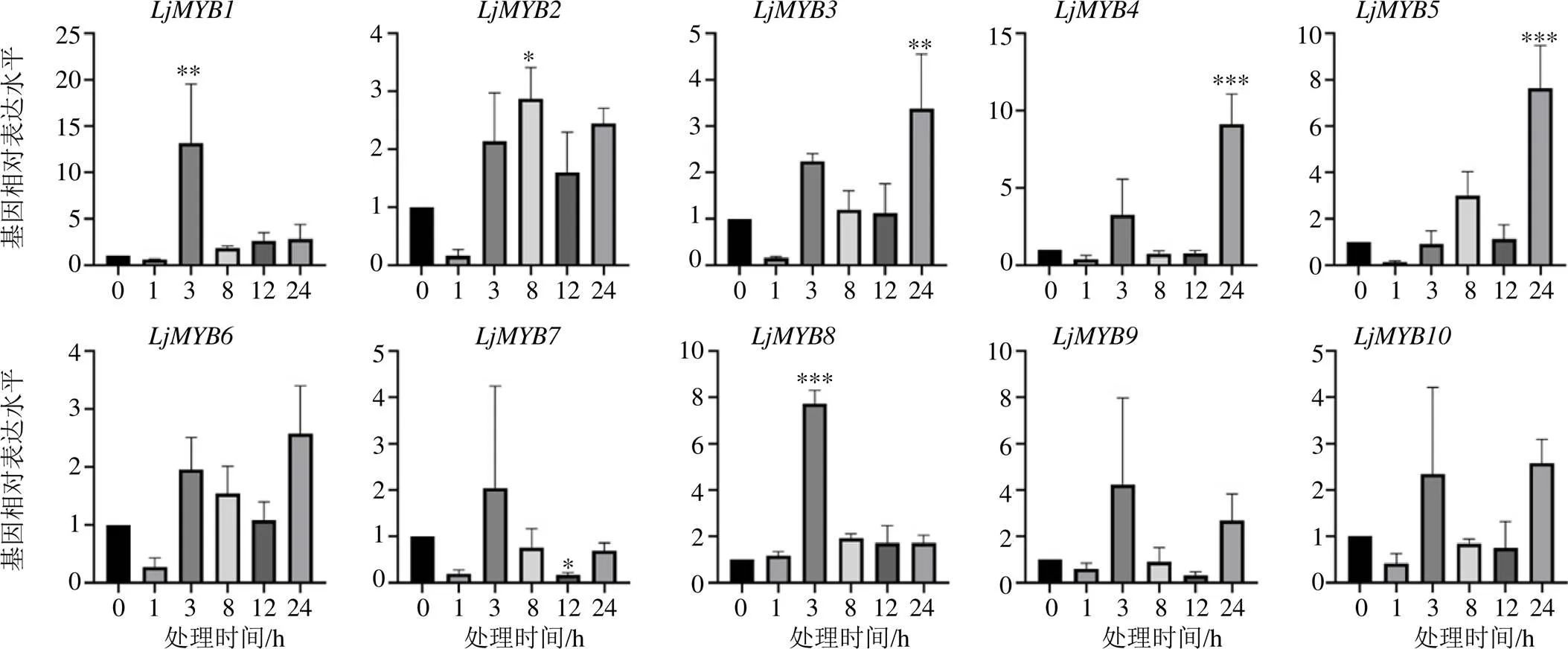
*P<0.05,**P<0.01,***P<0.001
有研究表明DNA甲基化在植物干旱胁迫响应中具有重要作用,Wang等[19]研究结果表明干旱诱导的DNA甲基化变化对水稻植株的干旱胁迫响应有相当大的影响,干旱诱导的DMRs在DK151植物中检测到更多,即在干旱条件下,甲基组在耐旱基因型中比在干旱敏感基因型中更稳定;Li等[20]采用WGBS法测定桑树在干旱胁迫下的甲基化变化发现,干旱胁迫后桑树甲基化水平普遍高于对照。本研究发现在干旱胁迫处理后,忍冬基因组甲基化水平普遍上调,且随着干旱胁迫时间的延长,甲基化水平呈上升趋势。进一步分析发现,在整体甲基化水平上升的同时,仍有部分基因甲基化水平下降,结合转录组数据进行分析,发现甲基化水平下降的差异甲基化基因其基因表达水平大部分也发生变化。富集分析显示这些基因主要富集于次生代谢产物合成相关通路,与忍冬中类黄酮类、萜类成分的合成密切相关。由此推测DNA甲基化调控基因表达是干旱影响金银花有效成分生物合成的作用机制之一。近年来,越来越多的研究表明,合成和积累次生代谢产物是药用植物最重要的防御环境胁迫的策略,环境胁迫导致药用植物次生代谢产物的积累,次生代谢产物通常也是药用植物的主要药用成分[21-22],通过本研究将为进一步探究环境影响金银花质量的分子机制奠定基础。
MYB转录因子是目前研究较多的转录因子家族,广泛参与调控植物的生长发育、抗逆胁迫、次级代谢产物的积累[23]。研究发现MYB转录因子的甲基化水平影响功能基因的表达进而对活性成分产生影响。通过基因组重测序结合转录组分析定量性状定位,发现红肉萝卜L. RsMYB1启动子区域的DNA高甲基化抑制基因表达,使花青素的生物合成收到抑制,导致了白色果肉表型[24]。Li等[25]研究发现MYB转录因子MdLUX和MdPCL启动子区mCG环境的甲基化水平与其mRNA水平和花青素积累呈负相关,MdLUX和MdPCL-like通过启动子区DNA低甲基化和结构类黄酮基因的激活促进苹果果皮花青素生物合成。Tang等[26]对异色菊花(YP)无性繁殖产生的黄花(YP-Y)和粉色花(YP-P)展开研究发现,CmMYB6启动子的甲基化水平是花色产生变异的关键,其甲基化程度影响着基因的表达和花青素的生物合成,对CmMYB6启动子进行去甲基化处理后,花朵颜色由黄色恢复为粉红色。这些研究显示MYB转录因子的甲基化水平对于植物体内类黄酮成分的生物合成有着重要作用。
类黄酮类成分是忍冬中的重要活性成分,是忍冬次生代谢产物的一种,前期有研究表明,适当的干旱胁迫会使类黄酮类成分含量增加[27],本研究通过组学分析加荧光定量PCR分析初步筛选出4个在干旱胁迫下表达量差异显著的转录因子,并通过通路富集分析发现其与忍冬的次生代谢产物的生物合成相关,下一步将继续深入探究这4个转录因子启动子区域甲基化的变化以及不同干旱胁迫处理下忍冬中活性成分的变化,为进一步解析忍冬在干旱胁迫下类黄酮类成分变化分子机制提供基础。
利益冲突 所有作者均声明不存在利益冲突
[1] 中国药典[S]. 一部. 2020: 230.
[2] 黄璐琦, 郭兰萍, 胡娟, 等. 道地药材形成的分子机制及其遗传基础 [J]. 中国中药杂志, 2008, 33(20): 2303-2308.
[3] 师凤华, 赵祥升, 莫乔程, 等. 不同产地不同种质金银花中绿原酸和木犀草苷的含量分析 [J]. 中药材, 2014, 37(7): 1145-1148.
[4] 徐迎春, 张佳宝, 蒋其鳌, 等. 水分胁迫对忍冬生长及金银花质量的影响 [J]. 中药材, 2006, 29(5): 420-423.
[5] 孙希芳, 王斌, 张永清, 等. 光照对金银花药材化学成分的影响 [J]. 山东农业科学, 2014, 46(10): 63-67.
[6] 王玲娜, 张金, 刘红燕, 等. 金银花质量影响因素分析 [J]. 中国医药导报, 2014, 11(25): 159-161.
[7] 张萍, 蒲高斌. 气候因子对金银花绿原酸含量影响的研究 [J]. 山东农业科学, 2015, 47(9): 77-79.
[8] 吕芳, 苏幼红, 张富春, 等. 植物活性成分对表观遗传调节的研究概况 [J]. 中草药, 2008, 39(10): 1580-1583.
[9] 薛梅, 陈成彬, 陈力, 等. 半夏多倍体复合体基因组DNA甲基化状态的MSAP分析 [J]. 中草药, 2008, 39(11): 1713-1716.
[10] Kuo T C, Chen C H, Chen S H,. The effect of red light and far-red light conditions on secondary metabolism in agarwood [J]., 2015, 15: 139.
[11] 谢宜杰, 刘晓莹, 罗可可, 等. 醇型和酮型广藿香DNA甲基化的MSAP分析 [J]. 中草药, 2020, 51(20): 5293-5301.
[12] Li L Q, Li X L, Fu C H,. Sustainable use of Taxus media cell cultures through minimal growth conservation and manipulation of genome methylation [J]., 2013, 48(3): 525-531.
[13] Zha L P, Liu S, Liu J,. DNA methylation influences chlorogenic acid biosynthesis in Lonicera japonica by mediating LjbZIP8 to regulate phenylalanine ammonia-lyase 2 expression [J]., 2017, 8: 1178.
[14] Pu X D, Li Z, Tian Y,. The honeysuckle genome provides insight into the molecular mechanism of carotenoid metabolism underlying dynamic flower coloration [J]., 2020, 227(3): 930-943.
[15] 王潇, 尹天舒, 李柏逸, 等. 基因功能富集分析的研究进展 [J]. 中国科学: 生命科学, 2016, 46(4): 363-373.
[16] 邢朝斌, 张妍彤, 王卓, 等. DNA甲基化及重亚硫酸盐测序法在药用植物中的应用策略 [J]. 中草药, 2017, 48(24): 5063-5069.
[17] Niederhuth C E, Bewick A J, Ji L X,. Widespread natural variation of DNA methylation within angiosperms [J]., 2016, 17(1): 194.
[18] 申晓慧. 两种诱变处理下紫花苜蓿全基因组DNA甲基化图谱分析 [J]. 草地学报, 2022, 30(1): 46-54.
[19] Wang W S, Qin Q, Sun F,. Genome-wide differences in DNA methylation changes in two contrasting rice genotypes in response to drought conditions [J]., 2016, 7: 1675.
[20] Li R X, Hu F, Li B,. Whole genome bisulfite sequencing methylome analysis of mulberry () reveals epigenome modifications in response to drought stress [J]., 2020, 10(1): 8013.
[21] 苏琦, 张珂, 黄淑琪, 等. 逆境胁迫对药用植物药效成分积累的影响 [J]. 湖北民族大学学报: 自然科学版, 2022, 40(2): 129-134.
[22] 郭兰萍, 周良云, 康传志, 等. 药用植物适应环境胁迫的策略及道地药材“拟境栽培” [J]. 中国中药杂志, 2020, 45(9): 1969-1974.
[23] 王晓童, 向丽, 邬兰, 等. 黄芩MYB转录因子家族全基因组鉴定与分析 [J]. 世界中医药, 2022, 17(13): 1802-1812.
[24] Wang Q B, Wang Y P, Sun H H,. Transposon-induced methylation of the RsMYB1 promoter disturbs anthocyanin accumulation in red-fleshed radish [J]., 2020, 71(9): 2537-2550.
[25] Li W F, Ning G X, Zuo C W,. MYB_ SH[AL]QKY[RF]transcription factors MdLUX and MdPCL-like promote anthocyanin accumulation through DNA hypomethylation and MdF3H activation in apple [J]., 2021, 41(5): 836-848.
[26] Tang M W, Xue W J, Li X Q,. Mitotically heritable epigenetic modifications of CmMYB6 control anthocyanin biosynthesis in chrysanthemum [J]., 2022, 236(3): 1075-1088.
[27] 岳凯, 刘文瑜, 魏小红. 干旱胁迫对不同品系藜麦内黄酮和抗氧化性的影响 [J]. 分子植物育种, 2019, 17(3): 956-962.
Whole genome DNA methylation and transcriptome analysis ofin response to drought stress
XU Xiao-han1, TANG Zhi-qiang1, LIU Qian1, 2, LI Jia1, 2, LIU Zhen-hua1, 2, ZHANG Yong-qing1, 2, PU Gao-bin1, 2
1. Shandong University of Traditional Chinese Medicine, Jinan 250355, China 2. Shandong Provincial Collaborative Innovation Center for Quality Control and Construction of the Whole Industrial Chain of Traditional Chinese Medicine, Jinan 250355, China
To analyze the changes and patterns of genome-wide DNA methylation level and its relationship with gene expression inunder drought stress, and lay a foundation for further exploring the molecular mechanism ofin response to drought stress.The whole-genome bisulfite sequencing (WGBS) was used to determine the DNA methylation level ofunder drought stress, and the differentially expressed genes related to differentially methylated genes were analyzed in combination with transcriptome sequencing results.The overall methylation level ofunder drought stress generally increased; The methylation level of CG type was significantly higher than that of CHG or CHH type. Differentially methylated regions (DMR) analysis ofunder different drought stress treatments revealed 1935, 2158 and 1750 differentially methylated genes. Kyoto encyclopedia of genes and genomes (KEGG) enrichment analysis showed that the significantly enriched pathways included linoleic acid metabolism, nitrogen metabolism and biosynthesis of secondary metabolites. Through transcriptome sequencing technology, it was found that differentially expressed genes related to differentially methylated genes were mainly enriched in secondary metabolite synthesis-related pathways, and fourtranscription factors with significant increase in gene expression under drought stress were screened.Under drought stress, the methylation level ofincreased, suggesting that DNA methylation regulation of gene expression is one of the mechanisms of drought affecting the biosynthesis of effective components of.
Thunb.; drought stress; DNA methylation; whole genome bisulfite sequencing; transcriptome sequencing
R286.12
A
0253 - 2670(2023)16 - 5339 - 11
10.7501/j.issn.0253-2670.2023.16.023
2023-02-03
山东省重点研发计划(乡村振兴科技创新提振行动计划)项目(2022TZXD0036);山东省农业良种工程项目(2021LZGC008);山东省现代农业产业技术体系中草药创新团队项目(SDAIT-20)
许小涵(1999—),女,硕士研究生,研究方向为中药分子生物学。Tel: 15194181753 E-mail: xuxiaohan0719@163.com
蒲高斌(1979—),男,博士,教授,主要从事中药资源与分子生药学研究。E-mail: gbpu@163.com
[责任编辑 时圣明]
猜你喜欢
新民周刊(2022年27期)2022-08-01
今日农业(2021年11期)2021-08-13
传染病信息(2021年6期)2021-02-12
生物医学工程学进展(2015年1期)2015-02-28
化学工业与工程(2015年1期)2015-02-10
现代检验医学杂志(2015年2期)2015-02-06
沈阳医学院学报(2014年4期)2014-12-27
遗传(2014年3期)2014-02-28
世界科学(2014年8期)2014-02-28
中国海洋大学学报(自然科学版)(2014年12期)2014-02-28
