
2,4,5- 三氨基-6- 羟基嘧啶硫酸盐中氰乙酸甲酯的方法研究
2023-08-08 10:21焦银蕾张晓灿何晓晨贾树红刘立平
煤炭与化工 2023年6期
焦银蕾,张晓灿,何晓晨,贾树红,刘立平
(河北冀衡药业股份有限公司 河北 衡水 053000)
0 引 言
叶酸是人体细胞生长和繁殖的必需物质,对人体的新陈代谢起着重要作用,是人体不可缺少的营养素之一,叶酸对细胞的分裂生长及核酸、氨基酸、蛋白质的合成起着重要的作用。但是由于天然的叶酸极不稳定,易受阳光、加热的影响而发生氧化,所以真正能从食物中获得的叶酸并不多,而合成的叶酸在数月或数年内保持稳定,容易吸收且人体利用度高。叶酸合成过程使用到原料为2,4,5- 三氨基-6- 羟基嘧啶硫酸盐(氨二)、对氨基苯甲酰谷氨酸、三氯丙酮。
2,4,5- 三氨基-6- 羟基嘧啶硫酸盐(俗称氨二),分子式:C4H9N5O5S,相对分子质量:239.21,CAS No.为35011-47-3,为生产原料药叶酸的主要使用原料,其制备过程是将氰乙酸甲酯、甲醇钠和硝酸胍混合在一起进行缩合反应,通过添加亚硝酸钠对混合物进行硝化,然后还原,最后加入硫酸,得到2,4,5- 三氨基-6- 羟基嘧啶硫酸盐(氨二),在其生产过程中使用氰乙酸甲酯作原料,故在反应不完全的情况下会有残留,从而影响原料药叶酸的产品质量,造成残留溶剂含量不合格。氰乙酸甲酯为无色至微黄色透明液体,可燃,为极性有机溶剂,不溶于水,可与乙醇、乙醚混溶。具有可燃性,有中等毒性,能经皮肤吸收,可引起皮肤炎症,需对其进行控制,目前各国药典均无相关检测方法,参考工业用氰乙酸甲酯国家标准GB/T26606-2011,采用气相色谱仪进行方法的研究,确定检测条件,制定质量标准,控制叶酸产品质量。
1 实 验
采用气相色谱仪进行分析的方法称为气相色谱法,气相色谱法是用气体作为流动相的色谱法,该方法分析速度快、分离效率高。
气相色谱法的原理是:样品溶液进样后,首先进入汽化室,然后在载气的传送作用下进入色谱柱(载气常用氮气或氦气),不同组分在色谱柱中被分离,最后依次流出色谱柱,被检测器检测,得到其含量。测定样品中某组分的含量时,需要先分析已知浓度的标准样品,然后将标准样品色谱峰的保留时间和峰面积与待测样品中改该组分比对,计算待测样品中目标组分含量。由于样品在气相中传递速度快,因此样品组分在流动相和固定相之间可以很快达到平衡,气相色谱法具有分析灵敏度高、检测时间短、应用范围广等优点。
本实验方法,需要确定检测方法能够将主成分和残留杂质及使用的溶剂进行有效分离,获得预期峰型,并达到分离度满足要求,定量限符合控制要求的目的。
1.1 试验用分析仪器及试剂:
主要仪器:分析天平(赛多利斯,型号QUIN IX124-1CN)、气相色谱仪(PE 气相色谱仪型号Clarus 580)、SPN-300 高纯度氮气发生器(北京中惠普分析技术研究所)、SPH-300 高纯度氢气发生器(北京中惠普分析技术研究所),SPB-3 全自动空气源(北京中惠普分析技术研究所)。
试剂:氰乙酸甲酯(上海麦克林生化科技有限公司)、无水甲醇(德国默克)
1.2 试验条件
1.2.1 确定色谱参数
2,4,5- 三氨基-6- 羟基嘧啶硫酸盐(氨二) 中残留氰乙酸甲酯能检测到,并且能够达到分离要求。初步确定的色谱参数为:
色谱柱:AE.SE-54 30m*0.32mm*0.25um;柱号:S.N.03111336001;检测器:FID;载气:氮气;载气流速:2.0 ml/min;进样量:0.5μl;检测器温度:240 ℃;柱箱:150 ℃;量程:1;分流比:20∶1;进样口温度:240 ℃。
1.2.2 溶液配制
空白溶剂:无水甲醇
对照品溶液的制备:精密称取氰乙酸甲酯对照品0.1 ml 于10 ml 容量瓶中,加入无水甲醇溶液至刻度,摇匀。
1.2.3 测定
吸取0.5μl 无水甲醇注入气相色谱仪中,作为空白溶剂;吸取对照品溶液0.5μl,注入气相色谱仪中,分别记录色谱图。
1.2.4 结果分析
发现对照品溶液中氰乙酸甲酯峰保留时间为1.566 min,溶剂峰保留时间为1.194 min,放大图谱后发现氰乙酸甲酯峰型出现峰前延现象,峰型不好,可能包含未分离的杂质,色谱图如下所示:图1 为完整谱图,图2 为放大后图谱。

图1 氰乙酸甲酯峰型图(保留时间1.566),完整图谱Fig.1 Methyl cyanoacetate Sample solution chromatogram(1.566),Complete map
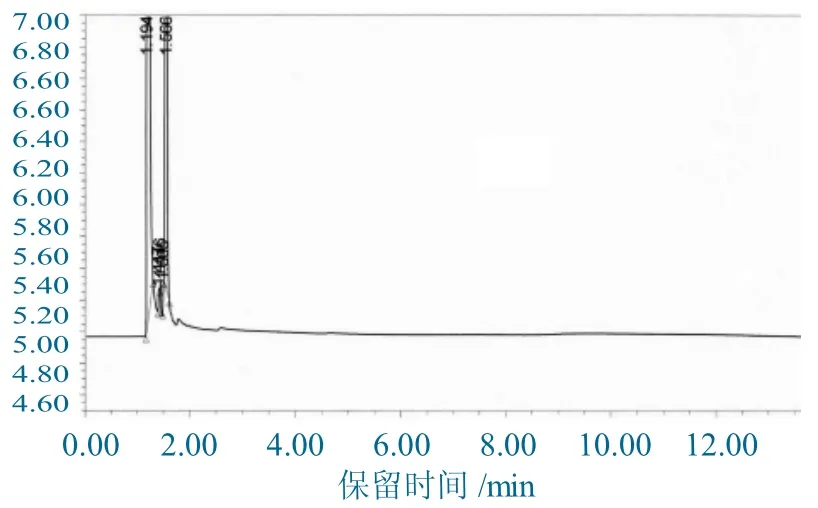
图2 氰乙酸甲酯峰型图(保留时间1.566),放大后图谱Fig.2 Methyl cyanoacetate Sample solution chromatogram(1.566),Magnification map
1.2.5 条件调整
其他条件不变,调整分流比:5∶1,再次进样。
发现对照中氰乙酸甲酯杂质峰保留时间为1.600 min,溶剂峰保留时间为1.204 min,和溶剂峰较近,放大图谱后发现氰乙酸甲酯峰型仍不理想。色谱图如图3 所示。

图3 调整分流比图谱(对照保留时间1.600)Fig.3 Chromatogram after adjusting Separation ratio(Retention time of reference solution 1.600)
1.3 优化氰乙酸甲酯峰型
1.3.1 调整色谱条件能够达到分离要求
文献表明,氰乙酸甲酯属于极性有机物质,AE.SE-54 柱子属于弱极性柱子,故可能存在杂质峰分离不开的状况,参照国标GB-/T26606-2011工业用氰乙酸甲酯的要求,我们更换检验参数及色谱柱。
调整柱温及程序升温:柱温是一个重要的操作变数,直接影响分离效能和分析速度。选择柱温是根据混合物的沸点范围,提高柱温可缩短分析时间;降低柱温可使色谱柱选择性增大,有利于组分的分离和色谱柱稳定性提高,色谱柱寿命延长。一般采用等于或高于数十度于样品的平均沸点的柱温较为合适,对易挥发样用低柱温,不易挥发的样品采用高柱温。对于沸点分布范围宽的多组分混合物,使用恒柱温气相色谱法分析,其低沸点组分会很快流出,峰形窄且易重叠,而高沸点组分则流出很慢,峰形扁平且拖尾,不利于定量测定还会延长分析时间。若使用程序升温气相色谱法,使色谱柱温度从低温开始,按一定升温速率升温,柱温呈线性增加直至终止温度,就会使混合物中的每个组分都在最佳柱温(保留温度)下流出。此时低沸物和高沸物都可在较佳分离度下流出,它们的峰宽窄适宜,并缩短总分析时间。
经查氰乙酸甲酯沸点204-207 ℃,此次试验继续调整程序升温温度、检测温度和速率,更换色谱柱型号。
1.3.2 色谱条件
色谱柱:PEG-20M 30m*0.32mm*0.25um;检测器:FID;载气:氮气;载气流:2.0ml/min;进样量:0.5μl;检测器温度:240 ℃;柱箱:240℃;量程:1;分流比:20∶1;进样口温度:240℃;柱温:起始温度140 ℃保持10 min,再由20℃/min 速率升到240 ℃,保持10 min。
1.3.3 溶液配制
空白溶剂:无水甲醇
对照品的制备:精密称取氰乙酸甲酯0.1 ml 于10 ml 容量瓶中,加入无水甲醇溶液至刻度,摇匀,作为对照品溶液。
1.3.4 测定
吸取0.5μl 无水甲醇注入气相色谱仪中,作为空白溶剂;吸取对照品溶液0.5μl 注入气相色谱仪中。经查看图谱,发现对照中氰乙酸甲酯杂质峰保留时间为8.214 min,溶剂峰保留时间为1.322 min,峰型较好,能够有效分离,记录色谱图如图4 所示。

图4 氰乙酸甲酯峰型图(保留时间8.214)Fig.4 Methyl cyanoacetate Sample solution chromatogram(8.214)
1.3.5 结果分析
经查看图谱,发现对照中氰乙酸甲酯杂质峰保留时间为8.214 min,溶剂峰保留时间为1.322 min,放大图谱后发现氰乙酸甲酯峰型良好,无杂峰现象,色谱条件调整达到预期效果。
1.4 确定方法检测限、定量限
1.4.1 检测限定量限研究
为使杂质峰型更理想化,其他条件不变,我们再次调节色谱参数:起始温度160 ℃。
根据2,4,5- 三氨基-6- 羟基嘧啶硫酸盐(氨二) 检验需要,须对其进行GC 氰乙酸甲酯的残留量检测,此方案主要研究2,4,5- 三氨基-6- 羟基嘧啶硫酸盐(氨二) 中是否能检测出氰乙酸甲酯,并且能够达到分离要求。为确定检验最小浓度我们将其对照液稀释100 倍。
1.4.2 色谱条件
色谱柱:PEG-20M 30m*0.32mm*0.25um;检测器:FID;载气:氮气;载气流速:2.0 ml/min;进样量:0.5μl;检测器温度:240 ℃;柱箱:240℃;量程:1;分流比:20∶1;进样口温度:240℃;柱温:起始温度160 ℃保持10 min,再由20℃/min 速率升到220 ℃,保持10 min。
1.4.3 溶液配制
空白溶剂:无水甲醇
对照品溶液的制备:精密称取氰乙酸甲酯0.1 ml 于10 ml 容量瓶中,加入无水甲醇溶液至刻度,再次吸取0.1ml 于10ml 容量瓶中,加入无水甲醇溶液至刻度,摇匀。
1.4.4 测定
吸取0.5μl 无水甲醇注入气相色谱仪中,作为空白溶剂;吸取氰乙酸甲酯对照品溶液0.5μl,记录色谱图。
1.4.5 结果分析
经查看图谱,发现对照中氰乙酸甲酯杂质峰保留时间为4.540 min,溶剂峰保留时间为1.256 min,放大图谱后发现氰乙酸甲酯峰型理想,无杂峰、前延、包峰等现象。计算分离度为37.5,信噪比为4,符合检测要求,色谱如图5 所示。

图5 氰乙酸甲酯峰型图(保留时间4.540)Fig.5 Methyl cyanoacetate Sample solution chromatogram(4.540)
1.4.6 专属性研究
按照以上色谱条件进行专属性研究,将对照品氰乙酸甲酯和2,4,5 三氨基-6 羟基嘧啶硫酸盐(氨二) 样品混合后,是否仍能检测到氰乙酸甲酯对照品、得到预期峰型和分离效果。
对照溶液:吸取氰乙酸甲酯0.5 ml 于10 ml 容量瓶中,加入无水甲醇溶液至刻度,摇匀。供试品混合溶液:精密称取2,4,5 三氨基-6 羟基嘧啶硫酸盐(氨二) 1.0 g 于10 ml 容量瓶中,吸取0.1 ml对照溶液于同一个10 ml 容量瓶中,加入无水甲醇溶液至刻度,摇匀。
测定时分别吸取空白溶剂甲醇、氰乙酸甲酯对照品溶液和过滤后供试品混合溶液的上清液,各0.5μl 分别注入气相色谱仪,记录所得色谱如图6 所示,对照品保留时间4.523 min;按面积归一法计算,混合溶液2,4,5 三氨基-6 羟基嘧啶硫酸盐(氨二) 中氰乙酸甲酯≤0.5%范围内对照品和溶剂峰分离较好,保留时间稳定,方法参数确定。

图6 特异性色谱图(对照溶液保留时间4.523 min)Fig.6 Specificity chromatogram(Retention time of reference solution 4.523 min)
2 验证方法的有效性
为验证方法的有效性,连续进样三批,2,4,5三氨基-6 羟基嘧啶硫酸盐(氨二) 进行测定。检测条件如下。
色谱柱:PEG-20M 30m*0.32mm*0.25um;检测器:FID;载气:氮气;载气流速:2.0 ml/min;进样量:0.5μl;检测器温度:240 ℃;柱箱:240℃;量程:1;分流比:20:1;进样口温度:240℃;柱温:起始温度160 ℃保持10min,再由20℃/min 速率升到220 ℃,保持10 min。
2.1 试剂:无水甲醇。
2.2 对照品溶液的制备
精密吸取氰乙酸甲酯0.1 ml 于10 ml 容量瓶中,加入无水甲醇溶解并稀释至刻度,摇匀。
供试品溶液的制备:精密称取0.5 g 样品于10 ml 容量瓶中,用无水甲醇溶解并稀释至刻度;取上清液作为供试品溶液。
2.3 测 定
分别吸取空白溶剂(无水甲醇)、对照品溶液和供试品溶液各0.5μl 分别注入气相色谱仪,记录色谱图,图7 为对照溶液图,4.435min 为对照品峰,色谱图8 为样品图谱。因为样品中氰乙酸甲酯残留较少故样品图谱中只有溶剂峰未检测到氰乙酸甲酯。
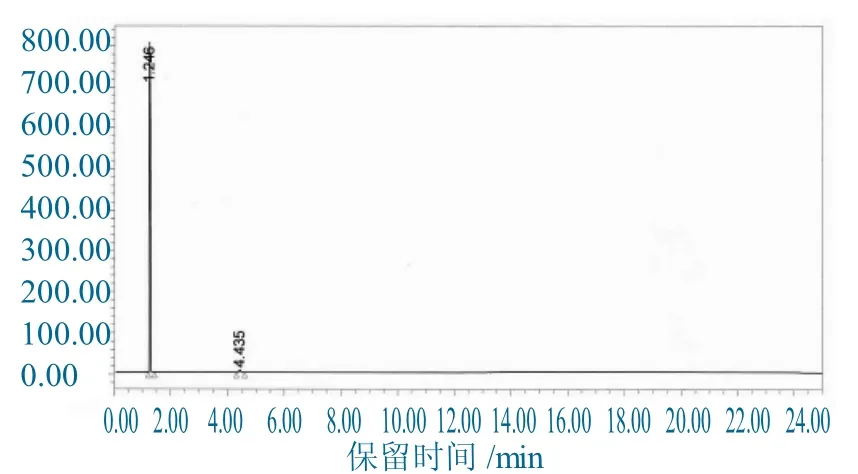
图7 对照溶液图谱Fig.7 Chromatogram of reference solution

图8 供试品溶液图谱Fig.8 Chromatogram of testing sample
2.4 计 算
按面积归一法计算,样品中残留溶剂氰乙酸甲酯≤0.5%。
3 结 语
经过调整参数,最终确定检测条件:以PEG-20M(30m*0.32mm*0.25um) 为色谱柱,FID检测器,载气流速:2.0 ml/min,进样量:0.5μl,检测器温度:240 ℃,量程:1,分流比:20∶1;在样品检测图谱中,对照品溶液中氰乙酸甲酯杂质峰保留时间为4.5 min 左右,溶剂峰保留时间为1.2 min 左右,分离度为37.5,信噪比为4,符合要求,初步确定2,4,5 三氨基-6 羟基嘧啶硫酸盐(氨二) 中氰乙酸甲酯残留限度为不得超过0.5%。
以上分析方法分离度、信噪比、峰型均能满足检测要求,分析方法及检测条件初步确定,该方法能准确可靠的测定2,4,5 三氨基-6 羟基嘧啶硫酸盐(氨二) 中残留氰乙酸甲酯的含量,为提高叶酸产品的质量提供了有效保障。
猜你喜欢
中学生数理化·高一版(2022年4期)2022-05-09
云南化工(2020年11期)2021-01-14
石油化工自动化(2018年5期)2018-11-14
浙江大学学报(工学版)(2016年9期)2016-06-05
现代工业经济和信息化(2016年4期)2016-05-17
当代化工研究(2016年5期)2016-03-20
化工进展(2015年3期)2015-11-11
中国当代医药(2015年10期)2015-03-01
海军医学杂志(2015年2期)2015-02-27
