
多药耐药蛋白1a对苯甲酰新乌头原碱的效-毒-体内暴露的调控研究
2023-08-05 08:01左慧琳李小翠区晓君杨彩华刘中秋梁奇朱丽君广州中医药大学中药学院国际中医药转化医学研究所广东广州10006中华人民共和国教育部中医药防治肿瘤转化医学研究联合实验室广东广州10006南方医科大学南方医院药学部广东广州101广州中医药大学粤港澳中医药与免疫疾病研究联合实验室广东广州10006深圳市宝安区中医院广东深圳181
中药新药与临床药理 2023年7期
左慧琳,李小翠,区晓君,杨彩华,刘中秋,4,梁奇,朱丽君,4(1. 广州中医药大学中药学院,国际中医药转化医学研究所,广东 广州 10006;2. 中华人民共和国教育部中医药防治肿瘤转化医学研究联合实验室,广东 广州 10006;. 南方医科大学南方医院药学部,广东 广州 101;4. 广州中医药大学粤港澳中医药与免疫疾病研究联合实验室,广东 广州 10006;. 深圳市宝安区中医院,广东 深圳 181)
苯甲酰新乌头原碱(Benzoylmesaconine,BMA)是中药附子(Aconiti Lateralis Radix Praeparata)的6 种质量控制标志物之一,具有显著的镇痛和抗炎活性[1-4]。BMA 能显著抑制醋酸诱导的疼痛模型小鼠的扭体反应,增加反复冷应激大鼠的足压痛阈值,降低胶原诱导性关节炎大鼠足肿胀的程度和关节炎指数[5-7]。但是,BMA 也具有一定的神经和心脏毒性,安全窗窄,口服和静脉给药BMA 在小鼠上的半数致死量(LD50)分别为810 和21 mg·kg-1[1-2,8]。对26 批附子的含量测定发现,BMA 是附子6 种质量控制标志物中含量最高的成分,最高可达389 μg·g-1,平均含量为144 μg·g-1[8]。可见,BMA 对附子的整体药效和毒性有着重要贡献[9],因此,阐明BMA 药效和毒性的控制作用机制对保障附子的有效应用与安全性具有重要意义。
多药耐药蛋白(Multidrug resistance protein 1,MDR1)是三磷酸腺苷结合盒转运蛋白家族成员,在体内组织器官中广泛分布,是影响药物体内处置及毒效的关键因素之一[10-17]。课题组前期采用Caco-2细胞模型研究表明,BMA 的外排受MDR1 调控。在MDR1 抑制剂的作用下,BMA 在Caco-2 细胞上的表观渗透系数增加约7.41 倍,外排率下降约75%[18]。然而,MDR1 是否参与调控BMA 的药效、毒性以及体内暴露尚不清楚。因此,本研究采用外排转运蛋白基因敲除的Mdr1a-/-小鼠(小鼠abcb1a基因编码的蛋白Mdr1a 与人类ABCB1 基因编码的蛋白MDR1 同源)和野生型FVB 小鼠模型来考察Mdr1a 对BMA 药效、毒性及体内暴露的影响,为BMA 的后续药理和药动学研究以及附子的安全有效应用提供实验依据。
1 材料与方法
1.1 仪器与材料1290 超高效液相色谱仪、6460 三重四极杆质谱仪,美国Aglient 公司;5810R 冷冻高速离心机,德国Eppendorf 公司;BSA224S-CW 电子分析天平,德国Sartorius 公司;SPD1010-230 真空离心浓缩仪,美国Thermo Fisher Scientific 公司;ElixEssential15(UV)超纯水系统,美国Millipore 公司;Tissuelyser-24 全自动样品快速研磨仪,上海净信实业发展有限公司。苯甲酰新乌头原碱,成都曼思特生物科技有限公司,纯度≥98%,批号:MUST-15 012216;利血平(内标),上海阿拉丁公司,纯度≥98%,批号:WXBC9972V;双氯芬酸钠,上海源叶公司,批号:Y29S11C 126393;醋酸,上海阿拉丁公司,批号:J1 811172;角叉菜胶,美国Sigma-Aldrich 公司,批号:SLBW1046;异氟烷,深圳瑞沃德生命科学有限公司,批号:21 103001;S100B 钙结合蛋白B(S100B)和肌酸激酶(CK)ELISA 试剂盒,武汉华美生物工程有限公司,批号分别为:M25016967和M23 016966;Hank’s 平衡盐溶液(HBSS),美国Sigma-Aldrich 公司,批号为SLBL7680V;其他化学药品和试剂均为分析纯及以上级别。
1.2 动物Mdr1a-/-小鼠,均为99%以上FVB 小鼠遗传背景,购自上海南方模式生物研究中心。野生型FVB 小鼠由北京维通利华实验动物技术有限公司提供。所有小鼠均为SPF 级别,6~8 周龄,雄性,体质量约21~25 g,实验前在SPF 级饲养室[温度(25 ±2)℃,相对湿度(50 ± 5)%,12 h 暗/光循环]适应性饲养1 周。实验前小鼠均禁食不禁水10~12 h。本研究所进行的所有动物实验均经广州中医药大学伦理委员会批准, 动物实验伦理审查表编号:IITCM20180907。
1.3 BMA 对Mdr1a-/-和野生型FVB 小鼠的镇痛作用采用醋酸致小鼠疼痛扭体模型考察镇痛作用。Mdr1a-/-和野生型FVB 小鼠按体质量随机分为3 组,每组6 只。各组小鼠均按10 mL·kg-1体积灌胃给药,空白组给予生理盐水,阳性药组给予双氯芬酸钠50 mg·kg-1,BMA 组小鼠给予BMA 20 mg·kg-1。灌胃30 min 后,腹腔注射1%醋酸(约0.22 mL)。腹腔注射醋酸5 min 后,记录15 min 内小鼠的扭体反应次数(腹部凹陷、躯干和后肢伸展和臀部翘起均视为扭体反应)[19]。
1.4 BMA 对Mdr1a-/-和野生型FVB 小鼠的抗炎作用采用角叉菜胶诱导小鼠急性炎症模型考察抗炎作用[19]。Mdr1a-/-和野生型FVB 小鼠按“1.3”项下分组与灌胃。灌胃30 min 后,测量小鼠右后足跖厚度(即造模前爪足厚度)。然后将小鼠放入装有2.5%异氟烷的配套小室中,保持1 min 以使小鼠完全麻醉。之后,将小鼠从小室取出,将小鼠的头部和鼻子放入装有1.0%异氟烷和500 mL·min-1氧气的麻醉面罩中,维持麻醉。在小鼠右后足跖下组织皮下注射20 μL 2%角叉菜胶。角叉菜胶处理4 h 后,再次测量右后足跖厚度(即造模后爪足厚度)。按照以下公式计算小鼠足肿胀率:足肿胀率(%)=(造模后爪足厚度-造模前爪足厚度)/造模前爪足厚度×100%。
1.5 UHPLC-MS/MS 分析方法
1.5.1 色谱条件 超高液相色谱系统为Agilent 1290 infinity LC system; 色谱柱为 Waters ACQUITY UHPLC HSS®T3 1.8 μm(2.1 mm×100 mm);流动相A 为0.1%甲酸-水溶液(甲酸∶超纯水=1∶999,V/V),流动相B 为乙腈;柱温为35 ℃;流速为0.4 mL·min-1,后运行1.00 min;进样室温度为6 ℃;进样体积为5 μL;洗脱程序为:0~1 min,30% B;1~2.5 min,30%~90% B;2.5~3.5 min,90% B;3.5~4.5 min,90%~30% B。
1.5.2 质谱条件 使用Agilent 6460 三重四级杆质谱,离子源为AJS ESI,采用正离子模式,多重反应监测(MRM)模式采集数据;毛细管电压为4.0 kV;干燥气温度为300 ℃;雾化器压力45 psi(1 psi≈6.895 kPa);喷嘴电压为500 V;鞘气温度为350 ℃;氮气体积流量为5 L·min-1;鞘气体积流量为11 L·min-1;定量分析的离子对:BMA(m/z590→540),利血平(m/z609→195);碎裂电压:BMA 135 V,利血平210 V;碰撞能:BMA 34 V,利血平38 V。
1.5.3 方法学考察[20]对小鼠血浆和肝脏组织中BMA的定量分析方法进行专属性、线性范围、精密度和准确度、提取回收率和基质效应、稳定性考察。
1.6 BMA 对Mdr1a-/-和野生型FVB 小鼠的神经和心脏毒性研究
1.6.1 脑和心脏的组织病理学观察 将Mdr1a-/-和野生型FVB 小鼠随机分为2 组,每组4 只。空白对照组灌胃生理盐水10 mL·kg-1,实验组灌胃BMA 20 mg·kg-1。给药8 h 后,处死小鼠,迅速取出脑和心脏,置于4%多聚甲醛中固定48 h 后,石蜡包埋,在冠状面切取厚度为6 μm 切片[19]。苏木精伊红染色,在显微镜下观察。
1.6.2 脑S100B 和心脏CK 水平的检测 动物分组和给药方法同“1.6.1”项。小鼠处死后迅速取出脑和心脏,分成两部分,一部分用于生化指标分析,另一部分用于BMA 的含量测定。用于生化指标分析的组织置于10 倍组织质量的冷冻1 × PBS 中,然后用组织匀浆器在65 Hz 下匀浆2 min,5 000g,4 ℃离心5 min,取上清液并立即进行检测。严格按照试剂盒说明书检测脑中S100B 和心脏中CK 水平。
1.6.3 BMA 在小鼠脑和心脏组织中的含量测定 将用于含量测定的组织样品放入新的1.5 mL 离心管中,加入2 倍组织质量的冷冻生理盐水(pH=7.4)进行匀浆,匀浆条件同“1.6.2”项。每个样品匀浆5 次。之后,将组织匀浆保存于-20 ℃,备用。
1.7 BMA 在Mdr1a-/-和野生型FVB 小鼠体内的组织分布研究将Mdr1a-/-和野生型FVB 小鼠随机分为2 组,每组4 只。静脉注射BMA 1 mg·kg-1,分别在0.5 和2 h 时间点从小鼠眼眶后静脉丛取血约20 μL,之后颈椎脱臼处死,迅速取出胆囊、脑、心、肝、肾、小肠和结肠并称质量。将组织样品放入新的1.5 mL 离心管中,加入2 倍组织质量的冷冻生理盐水(pH=7.4)进行匀浆,匀浆条件同“1.6.2”项。每个样品匀浆5 次。然后,将组织匀浆液保存于-20 ℃,待处理。将血液样本在956g下离心5 min,将上清液转移到新的离心管中,并保存于-20 ℃,备用。
1.8 BMA 在Mdr1a-/-和野生型FVB 小鼠体内的药动学研究将Mdr1a-/-和野生型FVB 小鼠随机分为2 组,其中Mdr1a-/-小鼠每组4 只,FVB 小鼠每组6 只。Mdr1a-/-和野生型FVB 小鼠灌胃BMA 20 mg·kg-1或尾静脉注射BMA 1 mg·kg-1后,分别在0、5、10、20、30、60、120、240、360、480、720 min 等不同时间点,从尾静脉采集血样约20 μL。然后,将血液样本在956g下离心5 min,将上清液转移到新的离心管中,并保存于-20 ℃,备用。
1.9 BMA 在Mdr1a-/-和野生型FVB 小鼠肠道吸收研究将Mdr1a-/-和野生型FVB 小鼠随机分为2 组,每组4 只。各组小鼠均按3 mL·kg-1体积腹腔注射50%乌拉坦麻醉剂,待小鼠麻醉后,将其固定在恒温床上,保持体温正常稳定。然后用酒精消毒小鼠表面皮肤,并剃除腹部毛发,沿小鼠腹中线打开腹腔约2 cm 至膈膜下端,暴露肠组织。在十二指肠和结肠的两端插入聚乙烯管,用手术线固定好。插好管后用37 ℃的生理盐水缓慢冲洗肠道内容物。然后用生理盐水浸润纱布,覆盖小鼠肠组织表面,保持肠道的湿润。用空白HBSS 缓冲液以10 mL·h-1的流速冲洗十二指肠和结肠15 min,以清洗其内容物。更换含10 μmol·L-1BMA 的HBSS 缓冲液,预灌流30 min,以达到稳态吸收。然后,在60 min 内每隔15 min 从十二指肠和结肠下端的出口管收集灌流液。灌流结束后,剪开整个十二指肠和结肠,测量并记录其长度。按文献方法[21]对有效表观渗透系数(Peff)和排泄到灌流液中的BMA 含量进行检测和计算。
1.10 样品制备取血浆(10 μL)或组织匀浆样品(20 μL)与50%(V/V)乙腈水溶液(10 μL)混合均匀。然后,在血浆或组织匀浆样品中分别加入180 μL 或170 μL 乙腈(含有50 nmol·L-1利血平)。涡旋4 min后,20 800g离心10 min。然后,将上清液(160 μL)转移到新的离心管中,常温下真空干燥2 h 后,加入50%乙腈水溶液80 μL 复溶,20 800g,4 ℃,离心30 min。取上清液5 μL,采用Agilent UHPLC-MS/MS 分析样品。胆汁样本的处理与血浆样本相同。灌流液样本的处理:取灌流液10 μL,加入50%乙腈水溶液90 μL(含100 nmol·L-1利血平),涡旋3 min后,在4 ℃,20 800g条件下离心30 min,取上清进样,采用Agilent UHPLC-MS/MS 分析样品。
1.11 数据分析采用Win Nonlin 3.3 软件(美国Pharsight 公司)进行药动学分析。采用GraphPad prism 5.0 软件进行数据作图。使用SPSS 20.0 统计学软件进行统计学分析。采用Student’st检验(两组)或One-way ANOVA 以及LSD 检验(两组或多组)评估显著性差异。以P<0.05 为差异有统计学意义。
2 结果
2.1 BMA 对Mdr1a-/-和野生型FVB 小鼠的镇痛作用结果见图1。在醋酸诱导的疼痛模型中,扭体次数越少表明镇痛效果越强。与空白对照组相比,FVB 小鼠和Mdr1a-/-小鼠口服BMA 后,扭体次数分别减少了55.09%和81.90%(P<0.05)。口服20 mg·kg-1BMA 后,与野生型FVB 小鼠相比,Mdr1a-/-小鼠的扭体次数下降60.82%(P<0.05)。

图1 苯甲酰新乌头原碱(BMA)对Mdr1a-/-和野生型FVB小鼠的镇痛作用(±s,n=6)Figure 1 The analgesic effects of benzoylmesaconine(BMA)on Mdr1a-/- and wild-type FVB mice(±s,n=6)
2.2 BMA 对Mdr1a-/-和野生型FVB 小鼠的抗炎作用结果见图2。在角叉菜胶诱导的急性炎症模型中,足肿胀率越低,抗炎作用越强。与空白对照组相比,FVB 小鼠和Mdr1a-/-小鼠口服BMA 后,足肿胀率分别减少了50.58%和74.34%(P<0.05)。与野生型FVB 小鼠相比,Mdr1a-/-小鼠的足肿胀率有下降趋势,但差异无统计学意义(P>0.05)。

图2 苯甲酰新乌头原碱(BMA)对Mdr1a-/-和野生型FVB小鼠的抗炎作用(±s,n=6)Figure 2 The anti-inflammatory effects of BMA on Mdr1a-/-and wild-type FVB mice(±s,n=6)
2.3 方法学考察小鼠血浆和肝匀浆中BMA 的代表性色谱图见图3。在BMA(1.6 min)和利血平(2.6 min)的保留时间处没有干扰组分引起的显著响应,分离效果较好,峰形理想。小鼠血浆和肝脏组织中BMA的线性范围、精密度、准确度、提取回收率、基质效应,以及稳定性考察结果见表1 ~表4。结果均显示该方法符合样品测定要求。

表1 苯甲酰新乌头原碱(BMA)在小鼠血浆和肝脏样品中的线性范围、回归方程、定量下限和相关系数Table 1 The linear range,regression equations,quantitative limit and correlation coefficients of BMA in plasma and liver of mice
表2 小鼠血浆和肝脏样品中苯甲酰新乌头原碱(BMA)日内和日间的精密度和准确度(±s,n=6)Table 2 Intra-day and inter-day precision and accuracy of BMA in plasma and liver of mice(±s,n=6)

表2 小鼠血浆和肝脏样品中苯甲酰新乌头原碱(BMA)日内和日间的精密度和准确度(±s,n=6)Table 2 Intra-day and inter-day precision and accuracy of BMA in plasma and liver of mice(±s,n=6)
日内/%浓度/(nmol·L-1)日间/%样品血浆肝脏9.77 312.50 5 000.00 19.54 312.50 5 000.00精密度(RSD)8.01 4.09 4.87 4.72 3.09 5.72准确度91.19±7.20 95.12±3.46 109.44±1.91 105.39±3.98 90.44±2.86 93.43±6.03精密度(RSD)6.88 4.60 2.59 4.41 4.61 7.84准确度99.75±8.23 100.56±4.04 107.20±3.59 104.12±5.74 90.70±3.86 94.02±6.15
表3 小鼠血浆和肝脏样品中苯甲酰新乌头原碱(BMA)的提取回收率和基质效应(±s,n=6)Table 3 The extraction recoveries and matrix effects of BMA in plasma and liver of mice(±s,n=6)
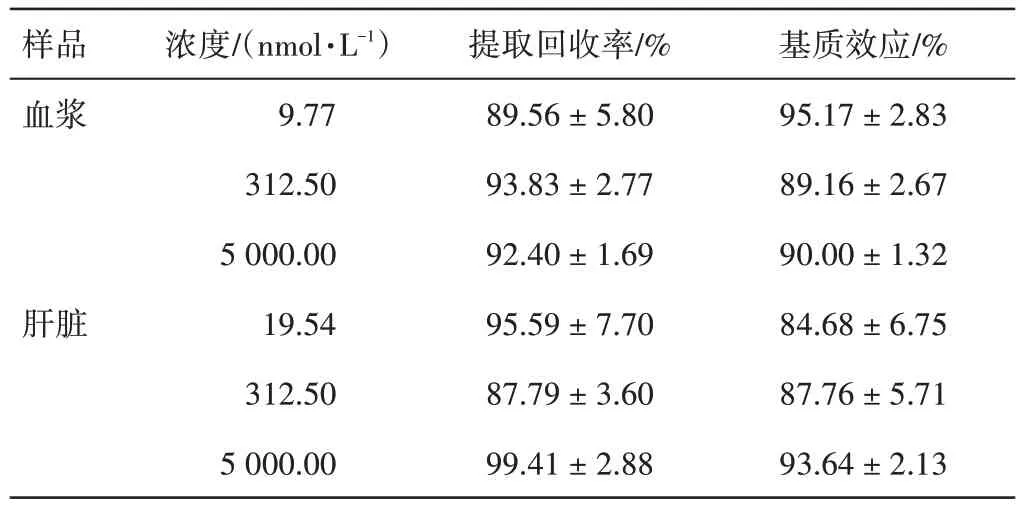
表3 小鼠血浆和肝脏样品中苯甲酰新乌头原碱(BMA)的提取回收率和基质效应(±s,n=6)Table 3 The extraction recoveries and matrix effects of BMA in plasma and liver of mice(±s,n=6)
样品血浆肝脏浓度/(nmol·L-1)9.77 312.50 5 000.00 19.54 312.50 5 000.00提取回收率/%89.56±5.80 93.83±2.77 92.40±1.69 95.59±7.70 87.79±3.60 99.41±2.88基质效应/%95.17±2.83 89.16±2.67 90.00±1.32 84.68±6.75 87.76±5.71 93.64±2.13
表4 小鼠血浆和肝脏样品中苯甲酰新乌头原碱(BMA)的稳定性(±s,n=6)Table 4 The stability of BMA in plasma and liver of mice(±s,n=6)

表4 小鼠血浆和肝脏样品中苯甲酰新乌头原碱(BMA)的稳定性(±s,n=6)Table 4 The stability of BMA in plasma and liver of mice(±s,n=6)
样品血浆肝脏浓度/(nmol·L-1)9.77 312.50 5 000.00 19.54 312.50 5 000.00室温放置6 h实测值/(nmol·L-1)10.22±0.82 318.06±12.58 5 473.29±120.44 20.33±0.56 283.29±11.65 4 784.57±306.07准确度/%104.65±8.39 101.78±4.03 109.47±2.41 104.12±2.88 90.65±3.73 95.69±6.12-20 ℃放置1 周实测值/(nmol·L-1)9.65±0.66 312.91±13.93 5 337.38±192.82 21.70±0.88 283.74±11.99 4 715.26±287.73准确度/%98.73±6.70 100.13±4.46 106.75±3.86 110.40±4.53 90.80±3.84 94.31±5.75-20 ℃至室温反复冻融3 次实测值/(nmol·L-1)9.29±0.77 311.75±12.78 5 269.28±178.32 21.77±1.28 282.25±14.63 4 603.89±353.58准确度/%95.08±7.87 99.76±4.09 105.39±3.57 111.98±6.57 90.64±4.68 92.08±7.07

图3 小鼠血浆和肝脏中苯甲酰新乌头原碱(BMA)的代表性色谱图Figure 3 Representative chromatograms of BMA in plasma and liver of mice
2.4 BMA 对Mdr1a-/-和野生型FVB 小鼠的神经毒性研究口服20 mg·kg-1BMA 后,Mdr1a-/-和野生型FVB 小鼠的脑组织切片、S100B 水平和BMA 含量测定结果见图4。口服BMA 8 h 后,Mdr1a-/-小鼠海马DG 区和CA 区细胞间隙增大(见图4-B2、-D2 图中黑色边框处)。与野生型FVB 小鼠相比,Mdr1a-/-小鼠口服BMA 后,海马区细胞数量减少,锥体细胞核固缩严重(图4-B2、-D2 图中黑色边框处)。与野生型FVB 小鼠相比,Mdr1a-/-小鼠脑中的S100B 水平和BMA 的含量分别增加了0.60 和3.12 倍(P<0.05)。

图4 苯甲酰新乌头原碱(BMA)的神经毒性及Mdr1a-/-和野生型FVB 小鼠脑内BMA 水平(±s,n=4)Figure 4 The neurotoxicity of BMA and BMA levels in the brain of Mdr1a-/- and wild-type FVB mice(±s,n=4)
2.5 BMA 对Mdr1a-/-和野生型FVB 小鼠的心脏毒性研究口服20 mg·kg-1BMA 后,Mdr1a-/-和野生型FVB 小鼠的心脏组织切片、CK 水平和BMA 含量测定结果见图5。口服BMA 8 h 后,与野生型FVB 小鼠相比,BMA 在Mdr1a-/-小鼠心脏中的分布增加了2.64 倍(P<0.05)。但各组小鼠心肌组织未见明显病理改变,心脏CK 水平也无明显变化。

图5 苯甲酰新乌头原碱(BMA)的心脏毒性及Mdr1a-/-和野生型FVB 小鼠心脏中BMA 水平(±s,n=4)Figure 5 The cardiotoxicity of BMA and BMA levels in the heart of Mdr1a-/- and wild-type FVB mice(±s,n=4)
2.6 BMA 在Mdr1a-/-和野生型FVB 小鼠组织中的分布特征结果见图6。与野生型FVB 小鼠相比,静脉注射1 mg·kg-1BMA 0.5 h 后,Mdr1a-/-小鼠脑、心脏、肾脏、结肠和血浆中BMA 的含量分别增加了1.06、1.34、0.59、2.36 和0.88 倍。给药2 h 后,与野生型FVB 小鼠相比,Mdr1a-/-小鼠脑、心脏、肾脏、小肠、结肠和血浆中的BMA 水平分别增加了1.58、0.67、1.27、2.08、1.22 和0.44 倍;胆汁中BMA的蓄积量减少约86.26%(P<0.05)。

图6 静脉注射1 mg·kg-1 苯甲酰新乌头原碱(BMA)0.5 和2 h 后,BMA 在Mdr1a-/-和野生型FVB 小鼠组织中的分布及在血浆和胆汁中的浓度(±s,n=4)Figure 6 Tissue distribution of BMA,the concentrations of BMA in the plasma and bile of Mdr1a-/- and wild-type FVB mice after intravenous injection of 1 mg·kg-1 BMA for 0.5 and 2 hours(±s,n=4)
2.7 BMA 在Mdr1a-/-和野生型FVB 小鼠体内的药动学特征BMA 在Mdr1a-/-和野生型FVB 小鼠体内的药代动力学特征和参数见图7 和表5。与FVB 小鼠相比,Mdr1a-/-小鼠体内BMA 的生物利用度(F)增加了4.53 倍(P<0.05)。同时,口服20 mg·kg-1BMA后,Mdr1a-/-小鼠体内BMA 的药时曲线下面积(AUC0-t)和AUC0-∞分别增加了1.46 倍和2.63 倍,清除率(Cl)和表观分布容积(Vd)分别降低了66.13%和78.85%(P<0.05)。静脉注射1 mg·kg-1BMA 后,与野生型FVB 小鼠相比,BMA 在Mdr1a-/-小鼠上的平均滞留时间(MRT0-∞)显著增加了1.79 倍(P<0.05)。
表5 Mdr1a-/-和野生型FVB 小鼠口服20 mg·kg-1 苯甲酰新乌头原碱(BMA)或静脉注射1 mg·kg-1 BMA 后的药动学参数(±s,n=4~6)Table 5 The pharmacokinetic parameters of BMA after oral administration of 20 mg·kg-1 or intravenous injection of 1 mg·kg-1 BMA in Mdr1a-/- and wild-type FVB mice(±s,n=4~6)

表5 Mdr1a-/-和野生型FVB 小鼠口服20 mg·kg-1 苯甲酰新乌头原碱(BMA)或静脉注射1 mg·kg-1 BMA 后的药动学参数(±s,n=4~6)Table 5 The pharmacokinetic parameters of BMA after oral administration of 20 mg·kg-1 or intravenous injection of 1 mg·kg-1 BMA in Mdr1a-/- and wild-type FVB mice(±s,n=4~6)
注:与野生型FVB 小鼠相比,*P<0.05
给药方式口服剂量/(mg·kg-1)20 Mdr1a-/-小鼠1.33±0.55*738.26±186.87 238.76±70.00*353.41±159.95*7.44±4.97 10.34±5.97 60.15±23.87*0.11±0.05*26.91±12.18*63.11±5.19 65.66±6.02 3.86±1.41 1.96±0.54*8.59±2.92 0.026±0.002 6参数静注1 tmax/h Cmax/(nmol·L-1)AUC0-t/(min·μmol-1·L-1)AUC0-∞(min·μmol-1·L-1)t1/2/h MRT0-∞/h Vd/(L·kg-1)Cl/(L·min-1·kg-1)F/%AUC0-t/(min·μmol-1·L-1)AUC0-∞/(min·μmol-1·L-1)t1/2/h MRT0-∞/h Vd/(L·kg-1)Cl/(L·min-1·kg-1)野生型FVB小鼠0.43±0.37 594.00±121.07 65.74±20.61 71.22±22.46 3.96±0.90 3.91±0.56 177.57±67.61 0.52±0.18 4.87±1.53 72.67±11.88 73.18±11.91 2.41±1.60 0.70±0.27 4.87±3.37 0.024±0.004 1

图7 小鼠单次灌胃20 mg·kg-1 苯甲酰新乌头原碱(BMA)(A)或静脉注射1 mg·kg-1 BMA(B)后的平均血药浓度-时间曲线(±s,n=4~6)Figure 7 The mean plasma concentration-time curves of BMA after oral administration of 20 mg·kg-1 BMA(A)or intravenous injection of 1 mg·kg-1 BMA(B)in Mdr1a-/- and wild-type FVB(±s,n=4~6)
2.8 BMA 在Mdr1a-/-和野生型FVB 小鼠肠道的吸收特征结果见图8。BMA 在小鼠十二指肠中的吸收特征数据显示,其有效表观渗透系数(Peff)从野生型FVB 小鼠的0.25 显著增加至Mdr1a-/-小鼠的1.25,即增加了4 倍(P<0.05)。与野生型FVB 小鼠相比,BMA 在Mdr1a-/-小鼠十二指肠的吸收率和吸收量均显著增加了3.96 倍(P<0.05)。Mdr1a 的缺失对BMA在小鼠结肠的Peff值、吸收百分率和吸收量无影响。

图8 苯甲酰新乌头源碱(BMA)在Mdr1a-/-和野生型FVB 小鼠肠道的吸收特征(±s,n=4)Figure 8 The intestinal absorption characteristics of BMA on Mdr1a-/- and wild-type FVB mice(±s,n=4)
3 讨论
苯甲酰新乌头原碱(BMA)是附子质量控制标志物之一,不仅具有显著的镇痛抗炎活性,还有一定的神经和心脏毒性[1-4]。前期研究[18]表明BMA 在体外被MDR1 外排,但MDR1 是否参与调控BMA 的镇痛抗炎活性、神经心脏毒性以及体内处置尚不清楚。人类ABCB1 基因编码的蛋白MDR1 与小鼠abcb1a 基因编码的蛋白Mdr1a 同源。本研究开展了BMA 对Mdr1a-/-和野生型FVB 小鼠的镇痛抗炎活性、神经心脏毒性以及药动学研究,发现Mdr1a 可通过显著改变BMA 在小鼠组织的分布、血浆暴露量和肠道吸收来调控其镇痛作用和神经毒性,但Mdr1a 对BMA 的抗炎作用和心脏毒性无明显调控作用。
本研究首先采用醋酸扭体模型和角叉菜胶诱导急性炎症模型来分别考察Mdr1a 对BMA 镇痛和抗炎活性的调控作用。结果发现BMA 的镇痛和抗炎作用较强,其作用强度与阳性药双氯芬酸钠相当。镇痛实验结果表明,BMA 在野生型FVB 小鼠上的疼痛抑制率约为55%(P<0.05)。据研究[5]报道,小鼠口服20 mg·kg-1BMA 后的疼痛抑制率约为52%,这与本研究结果基本一致。与野生型FVB 小鼠相比,Mdr1a-/-小鼠腹腔注射醋酸后,BMA 的镇痛作用明显增强,疼痛抑制率达到82%(P<0.05),Mdr1a 的缺失使BMA 的镇痛效果得以明显增强。然而,与野生型FVB 小鼠相比,BMA 对Mdr1a-/-小鼠的抗炎活性虽有增强趋势,但无显著性差异,这可能与角叉菜胶和醋酸的造模时间不同有关。本研究中醋酸作用5 min 即开始考察镇痛活性,而角叉菜胶需作用4 h才开始考察抗炎活性。由药动学结果可知Mdr1a-/-小鼠灌胃BMA 后,tmax为1.33 h,即4 h 前的时间点为BMA 的峰浓度(考察镇痛活性的时间在0.58~0.83 h之间),4 h 时间点后的血药浓度相对较低(考察抗炎活性的时间在4.5~4.6 h 之间)。这可能是Mdr1a 缺失会影响BMA 的镇痛作用但对抗炎作用无显著影响的原因之一。此外,这也可能与小鼠足跖下组织Mdr1a 的分布情况有关,Mdr1a 缺失是否会影响BMA在足跖下组织的分布尚不清楚。因此,后续研究可通过采用多种炎症模型,比如二甲苯诱导耳廓肿胀模型和棉球肉芽肿模型[22],以综合考察Mdr1a 对BMA 抗炎作用的调控机制。
本研究进一步采用组织病理切片和ELISA 法考察了Mdr1a 对BMA 神经和心脏毒性的调控作用。结果显示,口服BMA 8 h 后,与野生型FVB 小鼠相比,Mdr1a-/-小鼠的脑部海马DG 区和CA 区的细胞排列更加稀疏,细胞数量急剧下降。并且,Mdr1a-/-小鼠海马区出现明显的锥体细胞核固缩。S100B 是一种高度保守的钙结合蛋白,广泛分布于机体的中枢神经系统,在大脑发生严重损伤时,由大脑中的神经胶质细胞分泌,进入大脑内细胞外液,是判断大脑毒性程度的具体直观指标之一[23]。本研究结果发现,与野生型FVB 小鼠相比,Mdr1a-/-小鼠脑中的S100B水平显著增加(P<0.05)。同时,Mdr1a-/-小鼠脑中BMA 的浓度增加了3 倍。这证明了Mdr1a 可能通过调控BMA 在小鼠脑中暴露水平以增加其神经毒性。
野生型FVB 小鼠和Mdr1a-/-小鼠灌胃BMA 后,心肌纤维排列规则,形态结构正常,CK 水平也无明显变化。这表明Mdr1a 缺失对BMA 的心脏毒性无明显影响。这可能与心脏组织中外排转运蛋白的分布和功能有关。研究表明心脏组织表达的ABC 结合盒转运蛋白不仅参与药物以及心脏毒性物质的外排,还与心脏自身的离子通道传导特性和溶酶体功能密切相关[24]。同时,本研究仅考察了小鼠灌胃BMA 后心脏组织病理切片和CK 水平的变化,尚未对小鼠的心脏功能进行深入考察。因此,Mdr1a 缺失是否会影响BMA 的心脏毒性及其机制可能需要进一步研究。
本研究观察了Mdr1a 对BMA 在小鼠体内的组织分布和血浆药动学影响,并通过小鼠在体肠灌流实验进一步研究了Mdr1a 对BMA 肠吸收的调控作用。结果显示,静脉注射1 mg·kg-1BMA 2 h 后,与野生型FVB 小鼠相比,Mdr1a-/-小鼠胆汁中BMA 的蓄积量降低了86.26%(P<0.05)。与野生型FVB 小鼠相比,静脉注射1 mg·kg-1BMA 2 h 后,Mdr1a-/-小鼠脑、心脏、肾脏、小肠和结肠中BMA 浓度均显著升高(P<0.05)。与野生型FVB 小鼠相比,除消化器官外,BMA 的浓度在Mdr1a-/-小鼠中各器官的升高程度呈现脑>肾脏>心脏的趋势。与野生型FVB 小鼠相比,Mdr1a-/-小鼠静脉注射BMA 0.5 或2 h 后,脑和心脏中BMA 的浓度均显著上升,该结果与毒性研究中的BMA 含量测定结果一致。大脑是乌头类生物碱中毒的首要靶器官,中毒者常出现中枢神经系统毒性症状[25]。BMA 在Mdr1a-/-小鼠脑中积累的增加为其镇痛作用的增强和神经毒性的加剧提供了很好的解释。心脏不是MDR1 表达最为丰富的器官,MDR1主要分布在脑、小肠和肾脏中[16]。因此,BMA 在Mdr1a-/-小鼠心脏中的浓度增加程度小于其在脑组织中,这可能也解释了BMA 在心脏中的蓄积不会产生类似神经毒性那么明显的原因。
Mdr1a 也分布在肾脏的刷状缘膜上,对药物的肾脏排泄有一定的影响[16]。本研究结果表明,肾脏是BMA 的主要分布和排泄器官。BMA 在肾脏中的消除比在其他器官中更慢,尤其是在Mdr1a-/-小鼠中,与FVB 小鼠相比,BMA 在肾脏中的浓度明显升高,消除速度减慢。这一结果提示,当MDR1 在体内被抑制时,可能会增加BMA 致肾损伤的风险。此外,与野生型FVB 小鼠相比,Mdr1a-/-小鼠血浆中BMA 浓度显著升高,这是因为Mdr1a 外排转运的缺失,减少了BMA 外排至肠腔,从而增加了BMA 吸收入血。
在血浆药动学研究中,发现Mdr1a 外排转运蛋白的缺失显著增加了BMA 在小鼠体内的暴露量,并显著减缓了BMA 的消除速率。与野生型FVB 小鼠相比,BMA 在Mdr1a-/-小鼠体内的生物利用度增加4.53 倍,tmax、AUC0-t和AUC0-∞显著增加,Vd值和Cl显著减少(P<0.05)。作者认为Mdr1a 可能不仅仅通过影响BMA 的体内组织分布来影响BMA 的药动学过程,基于Mdr1a 在肠道中广泛分布的特性,Mdr1a还可能通过影响其吸收来影响BMA 的药代动力学过程。胃肠道吸收是口服药物发挥疗效的重要前提,而吸收部位包括胃、小肠和大肠,其中小肠的吸收最为重要。体内肠灌流模型是美国食品药品监督管理局(FDA)批准的用于研究药物吸收特性的肠吸收模型,该模型接近药物在体内的真实吸收状态[26]。在BMA 的肠道处置研究中,与FVB 小鼠相比,BMA 在Mdr1a-/-小鼠十二指肠中的吸收显著增加,Peff值增加了约4 倍,这一结果与血浆药动学实验结果一致(BMA 在Mdr1a-/-小鼠体内的生物利用度增加了约4.5 倍)。结合对肠灌流实验、药代动力学、药效和毒性实验结果的分析,Mdr1a 可通过影响BMA 的肠吸收来调节BMA 的体内暴露,进而调控其毒效。BMA 在Mdr1a-/-小鼠中吸收程度的增加,镇痛作用的增强和神经毒性的加剧,预示着可能引起具有临床意义的药效增加或不良反应发生[27-28]。
综上所述,Mdr1a 通过改变BMA 的组织蓄积、体内暴露量和肠道吸收,参与调节BMA 的镇痛作用和神经毒性。然而,BMA 的抗炎作用和心肌毒性可能与Mdr1a 的缺失无关,后续实验应建立多种炎症模型和应用心肌毒性评价指标综合验证Mdr1a 对BMA 抗炎作用和心肌毒性的影响。本研究在小鼠上证实了Mdr1a 对BMA 的药效和毒性的调控作用,为附子的质量控制、疗效发挥、安全用药和研发应用提供了实验基础,并提示使用附子时,应注意配伍药物是否为潜在的Mdr1a 抑制剂,但是关于BMA 与Mdr1a 抑制剂联合用药的药物-药物相互作用的研究有待进一步探讨。
猜你喜欢
中国饲料(2021年17期)2021-11-02
中华养生保健(2020年7期)2020-11-16
中成药(2018年11期)2018-11-24
中成药(2018年2期)2018-05-09
中成药(2017年9期)2017-12-19
中成药(2017年10期)2017-11-16
广东饲料(2016年5期)2016-12-01
中成药(2016年4期)2016-05-17
合成化学(2015年10期)2016-01-17
中药与临床(2015年5期)2015-12-17
