
Isolated IgG4-associated autoimmune hepatitis or the first manifestation of IgG4-related disease?
2023-08-02 09:00:48CarmelaCosentinoDanielClaytonChubbCatrionaMcLeanStuartRobertsWilliamKemp
Carmela Cosentino , Daniel Clayton-Chubb , Catriona McLean , Stuart K Roberts ,William Kemp
a Department of Gastroenterology and Hepatology, Alfred Health, Melbourne 3004, VIC, Australia
b Central Clinical School, Monash University, Melbourne 3004, VIC, Australia
c Department of Anatomical Pathology, Alfred Health, Melbourne 3004, VIC, Australia
TotheEditor:
Immunoglobulin G4 (IgG4)-associated autoimmune hepatitis(IgG4-AIH) is a novel and rare disease entity, characterized by significant infiltration of IgG4-expressing plasma cells in the liver.The classification of of IgG4-AIH as a subtype of AIH or an early manifestation of IgG4-related disease (IgG4-RD) remains controversial.Herein, we discuss an interesting clinical vignette of IgG4-AIH in a gentleman with no significant past medical history, who presented with undifferentiated symptoms and elevated aminotransferases.
A 58-year-old man was admitted to hospital with 3 weeks of progressive fatigue, nausea and deranged liver function tests.He denied any subjective fevers, altered bowel habit, recent viral illnesses, anorexia, or weight loss.There was no recent travel.His regular medications include venlafaxine, turmeric, vitamins C and D, glucosamine, and multivitamins with no recent changes to these or antibiotic use.There was no other significant past medical or surgical history or family history of liver, hepatobiliary or autoimmune diseases.He was a non-smoker and consumed one-to-two standard alcoholic drinks daily with no recreational drug use or at risk sexual behaviors.In addition, clinical examination was unremarkable.Of note, he was apyrexial with no evidence of scleral icterus.His abdomen was soft, non-tender and negative for Murphy’s sign.His liver was not enlarged.There were no clinical signs of chronic liver disease or splenomegaly.
The liver function test abnormalities included an alanine aminotransferase (ALT) of 1637 U/L (reference range ≤40 U/L), aspartate aminotransferase (AST) of 893 U/L (normal ≤35 U/L), total bilirubin of 32 μmol/L (normal ≤21 μmol/L), gamma-glutamyl transferase (GGT) of 221 U/L (normal ≤62 U/L) and alkaline phosphatase (ALP) of 130 U/L (normal range 30-110 U/L).His viral serology was negative for current or previous hepatitis A, B, C or HIV.He was immunoglobulin G (IgG) positive/immunoglobulin M (IgM)negative for both cytomegalovirus and Epstein-Barr virus.
An autoimmune screen yielded a positive homogeneous antinuclear antibody with a titer of 1:640, elevated serum IgG level of 27 g/L (normal range 7-16.5 g/L), and a marked elevation in its subclass IgG4 of 5.74 g/L (normal range 0.04-0.86 g/L).Anti-smooth muscle antibody (ASMA), anti-mitochondrial antibody(AMA), anti-liver kidney microsomal (anti-LKM), anti-soluble liver antigen (anti-SLA) and anti-liver cytosolic antigen-1 antibodies(anti-LC1) were all negative.Ceruloplasmin and alpha-1-antitrypsin(A1AT) were within normal limits.His ferritin was markedly elevated at 5422 μg/L with a transferrin saturation of 87%.
Additional investigations including abdominal Doppler ultrasound and magnetic resonance cholangiopancreatogram were both unremarkable, with no biliary tract stricturing and a normalappearing pancreas with no features of autoimmune pancreatitis(AIP) or IgG4-related sclerosing cholangitis (IgG4-SC).His serum lipase level of 43 U/L was also normal (normal range 10-70 U/L).Over the following 5 days his liver function tests continued to fluctuate without significant improvement ( Fig.1 ).
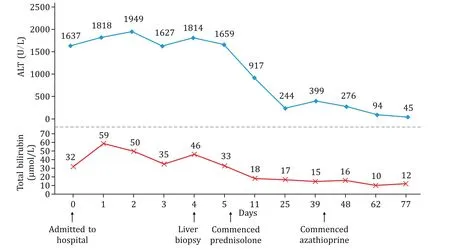
Fig.1.Liver function tests and treatment response.ALT: alanine aminotransferase.
A percutaneous core liver biopsy was conducted at day 4 of his admission.Histological examination of the core liver biopsy (20 mm in length with 12 portal tracts) demonstrated a moderately florid predominantly plasma cell infiltrate within the portal tracts and lobules and prominent interface hepatitis ( Fig.2 ), occasional eosinophils and normal bile ducts with an increase in portal tract fibrosis.The pre-treatment revised International Autoimmune Hepatitis Group (IAIHG) disease score [1]was 17, identifying definite AIH.In view of the elevated serum IgG4 level, immunostaining for IgG4 was performed which identified an IgG4 to IgG ratio of approximately 30% with some portal tracts having>10 IgG4 plasma cells per high-power field (HPF).A diagnosis was made of IgG4-AIH.The patient had symptomatic and biochemical improvement with a tapering course of prednisolone 40 mg daily followed by azathioprine maintenance therapy ( Fig.1 ).IgG levels were normalized (13.9 g/L) after 38 days of therapy.The18F-FDG positron emission tomography (PET) scan was performed which did not show any evidence of systemic or extra-hepatic IgG4 disease (IgG4-RD).
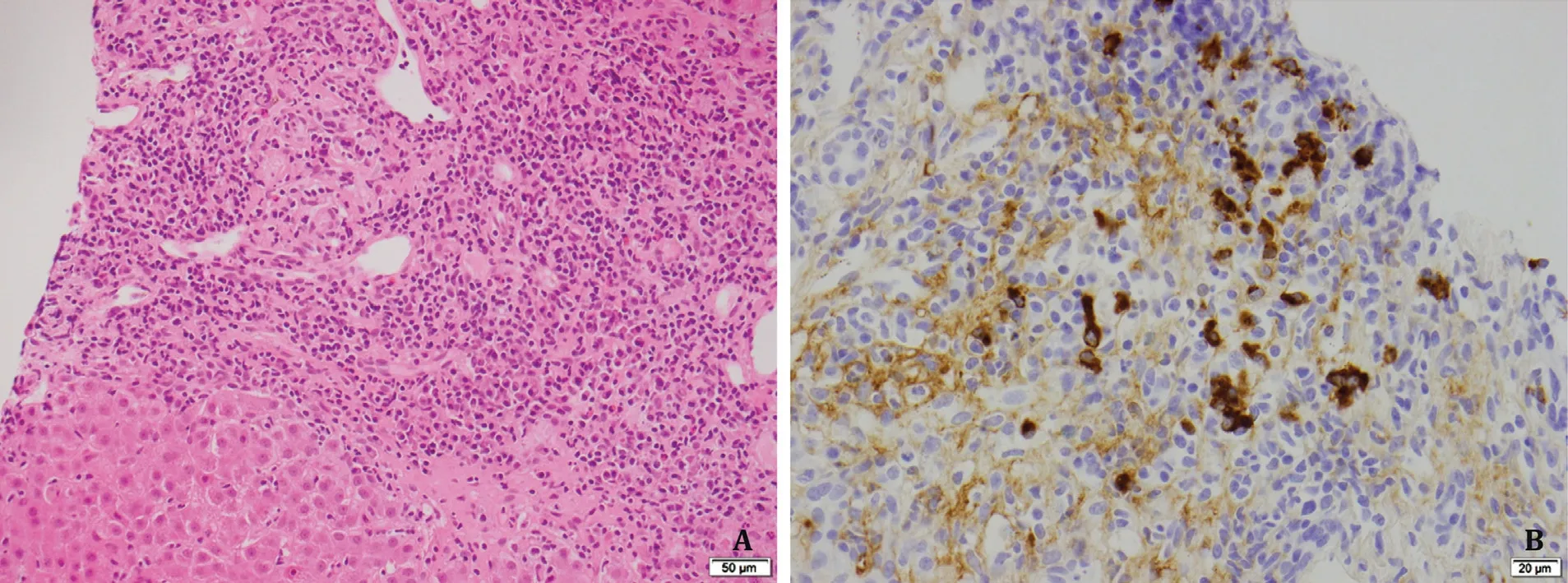
Fig.2.Histological liver biopsy findings and biochemical trends.A: Liver biopsy before therapy showing a concentrate of plasma cells in an expanded portal tract with interface change (hematoxylin and eosin staining, original magnification × 200).B: Immunostaining showing IgG4-bearing plasma cells (IgG4 immunoperoxidase staining,original magnification × 400).
Similar to our case, the original report of “IgG4 associated autoimmune hepatitis” in 2007 described a 54-year-old female with marked elevation of serum IgG4 and a plasma cell rich hepatic infiltrate with strong immunohistochemical reactivity to IgG4 and a good response to prednisolone [2].However, following recognition of this clinicopathological entity, the significance of an elevated serum IgG4 levels in conjunction with an abundant hepatic IgG4 plasma cell infiltrate remains uncertain.
While it is recognized that IgG4-positive plasma cells may be a histopathological feature of some patients with AIH [3], a review by Minaga et al.[4]provides context for the novel disease entity now referred to as IgG4-associated AIH (IgG4-AIH).Depending on the cut-off of IgG4 expressing plasma cells per HPF used to define IgG4-AIH (>5 IgG4 + cells/HPF vs.>10 IgG4 + cells/HPF), the proportion of classical AIH being re-classified as IgG4-AIH varies considerably from 3.3% to 34.6%.These histological findings suggest that a particular subtype of AIH may exist, characterized by significant infiltration of IgG4-expressing plasma cells in the liver.However, this subtype cannot be reliably distinguished by either biochemical or histological characteristics nor by treatment response.
Based on this definition, our case demonstrated features of classical AIH with>10 IgG4 + cells/HPF, fulfilling the criteria for IgG4-AIH.Interestingly, our case also exhibited a significant elevation in serum IgG4 level.The co-existence of these two features represents a much rarer phenomenon limited to a few case reports,with a review by Nakanuma et al.highlighting the existence of only 3 well described cases [5].Included in this report is a followup to the original 2007 Umemura et al’s case of IgG4-AIH [2]who interestingly developed features of IgG4-SC 5 years after her original presentation.Nakanuma et al.[5]put forward alternative diagnostic criteria for IgG4-AIH limiting the definition of “probable”and “definite” IgG4-AIH to those with elevated serum IgG4 and highlighting the potential for this disease entity to manifest additional metachronous or synchronous organ involvement of IgG4-RD.Moreover, these diagnostic criteria distinguish IgG4-AIH from other liver conditions with IgG4 plasma cell infiltrates such as IgG4-hepatopathy which is associated with type 1 AIP or IgG4-SC, “denovo” AIH associated with liver allografts, and hepatic inflammatory pseudotumor as a manifestation of IgG4-RD.Despite emerging evidence for IgG4-AIH as a distinct entity, the existence of IgG4-related hepatitis as a primary manifestation of IgG4-RD remains controversial and further characterization is required.Our case had no clinical or radiological features (including PET scan) of IgG4-RD at diagnosis or over the following five months of followup.However, longer-term vigilance will be necessary.
In this report, we reviewed the historical and recent literature describing the diagnosis, and nuanced management and follow-up of patients with IgG4-AIH, to support the education of clinicians managing patients with this rare condition.
Acknowledgments
None.
CRediT authorship contribution statement
Carmela Cosentino:Conceptualization, Data curation, Investigation, Visualization, Writing - original draft, Writing - review &editing.Daniel Clayton-Chubb:Conceptualization, Data curation,Investigation, Visualization, Writing - original draft, Writing - review & editing.Catriona McLean:Data curation, Investigation, Visualization, Writing - review & editing.Stuart K Roberts:Investigation, Writing - review & editing.William Kemp:Conceptualization, Investigation, Visualization, Writing - original draft, Writing -review & editing.
Funding
None.
Ethical approval
Written informed consent for publication was obtained from the patient.
Competing interest
No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
 Hepatobiliary & Pancreatic Diseases International2023年4期
Hepatobiliary & Pancreatic Diseases International2023年4期
- Hepatobiliary & Pancreatic Diseases International的其它文章
- Novel re-intervention device for occluded multiple uncovered self-expandable metal stent (with video)
- Hepatopancreatoduodenectomy for the treatment of extrahepatic cholangiocarcinoma ✩
- Microbiological cultures and antimicrobial prophylaxis in patients undergoing total pancreatectomy with islet cell autotransplantation
- Risk factors for posttransplant diabetes in patients with hepatocellular carcinoma
- The role of targeting protein for Xklp2 in tumorigenesis of hepatocellular carcinoma
- Left abdominal mass with carcinosis: Unusual presentation of pancreatic acinar cell carcinoma