
TpBD(NH2)2的D-葡萄糖衍生物高效液相色谱固定相研究*
2023-08-01 07:59李斗发李亚楠刘华林
云南化工 2023年7期
李斗发,李亚楠,刘华林,字 敏
(云南师范大学 化学化工学院,云南 昆明 650500)
引言
在临床上使用的很多药品是手性药物[1]。不同的异构体具有不同的生理活性、物理和化学性质,有些药品一个对映体有药效,另一个对映体可能没有药效,甚至对人的身体健康有害[2]。所以,对具有手性的药物进行分离具有重大的现实意义。由于外消旋体的理化性质差别很小,很难把对映异构体分离开[3],分离手性药物的方法很多,其中比较典型的有毛细管电色谱(SEC)、高效液相色谱(HPLC)、气相色谱(GC)等[4-6]。手性固定相CSP(Chiral stationary phase,CSP)分离手性化合物是当前比较热门的研究方向,使用比较广的是多糖、环糊精和冠醚作为固定相[7-9]。
在过去的十多年里,研究员对共价有机骨架材料COFs(covalent organic frameworks,COFs)做了很多探索和研究,因为其具有很多优良的性能,例如,具有高的表面积、孔的体积、形状容易调节,方便组装[10]。COFs大量应用于气相色谱、液相色谱[11]、催化和识别等[12]领域。
本文使用后合成修饰法[13]和溶剂热合成法[14],获得了具有手性的TpBD(NH2)2的D-葡萄糖衍生物,将其采用网包法[15-16]覆盖在球形硅胶上,用于制备手性固定相。实验结果表明TpBD(NH2)2的D-葡萄糖衍生物能够很好地分离手性药物、氨基酸以及位置异构体。与此同时,制备的手性固定相填充的分离柱具有较好的稳定性。通过研究表明,制备的手性CSP在分离手性化合物具有较大的潜能。
1 实验部分
1.1 试剂和仪器
D-Max-3B 2000X粉末衍射仪(日本,Rigaku公司);S-3000N扫描电子显微镜(日本,Hitachi公司);Chirascan圆二色光谱仪(英国,应用光学物理);BRUKERTensor-27傅里叶红外变换红外光谱仪(德国,Bruker);ASAP2020 氮气吸附仪(美国,Micromeritics公司);EliteP230Ⅱ高压流压泵(大连依力特分析仪器有限公司);EliteUV230Ⅱ 紫外检测仪(大连依力特分析仪器有限公司);AT-330柱温箱(大连依力特分析仪器有限公司);1666液相色谱装柱机(美国,Alltechi有限公司);200 mm×φ4.6 mm 不锈钢高效液相色谱空柱(美国,Alltechi有限公司);UniSil 5-100硅胶(苏州纳微科技股份公司)。
六亚甲基四胺(97%)、三氟乙酸(97%)、邻硝基苯胺(99%)、碘化钾(98%)、高碘酸钾(99%)、位置异构体(99%)均购于上海阿拉丁生化科技股份有限公司;三乙胺(≥99%)、二茂铁二氯化钯(99%)、均三甲苯(99%)、无水SnCl2(99%)均购于上海阿达玛斯试剂公司;氯化钠(≥99%)、葡萄糖(99%)购于天津市风船化学试剂科技有限公司;1,4二氧六环(98%),外消旋化合物(99%)均购于美国Sigma-Aldrich有限公司;间苯三酚(98%)购于郑州阿尔法化工有限公司。
1.2 单体1,3,5-三甲醛间苯三酚(Tp)的合成
取用经干燥后的 7.549 g 六亚甲基四胺(54 mmol),3.007 g 间苯三酚(24.5 mmol),加入 500 mL 的三颈烧瓶中。对装置抽真空后充满N2,在N2的保护下,边搅拌边加入 90 mL 三氟乙酸,后将装置移入油浴锅,在 100 ℃ 下搅拌 3 h 后,取用配制好的 0.3 mol/L 的盐酸溶液 150 mL 加入到反应容器中,再反应 2.5 h。烧瓶取出冷却后,用二氯甲烷萃取产物,取有机相层,蒸发掉溶液后,用甲醇抽滤洗涤产物,得到单体1,3,5-三甲醛间苯三酚(Tp),产物呈粉色。
1.3 单体3,3′-二硝基联苯胺(DNB)的合成
取 1.24 g 4-碘-2-硝基苯胺(5 mmol),三乙胺 1.4 mL,0.016 g 二茂铁二氯化钯(DPPF,0.5 mmol),17.5 mL 无水甲苯,依次加入到 100 mL 二颈烧瓶中,在90~130 ℃ 下搅拌反应 12 h,得到单体3,3′-二硝基联苯胺(DNB),产物呈红色、粉末状。
1.4 TpBD(NH2)2的合成
称取 21.0 mg 的Tp(0.1 mmol),41.1 mg 的DNB(0.15 mmol),均三苯甲醛 0.75 mL,1,4-二氧六环 0.75 mL,3 mol/L 醋酸 0.5 mL,于Pyrex耐热玻璃管中,超声至其混合均匀。用液氮冷冻后,抽真空脱气,用酒精喷灯封管,在 120 ℃ 的条件下反应 72 h,待反应完成后,将玻璃管敲碎,取出产物,依次用丙酮、二氯甲烷洗涤,得到呈红棕色的产物。
取 3 g SnCl2·2H2O和 150 mg 上述合成的TpBD(NO2)2于 50 mL 的二颈烧瓶中,加入 5 mL 无水四氢呋喃使其混合溶解,在 50 ℃ 条件下加热回流 3 h。反应完成后,将沉淀物用 70 mL 的 1 mol/L 盐酸洗涤10次,用 70 mL 的水洗涤3次,并用 100 mL 丙酮洗涤一次。然后取上步产物于合适的反应釜中,加入 5 mL 苯甲醚,在 120 ℃ 烘箱内加热 24 h。最后,将粉末过滤并用 100 mL 丙酮洗涤。得到红棕色产物 TpBD(NH2)2,合成路线如图1。

图1 TpBD(NH2)2的合成路线
1.5 TpBD(NH2)2的D-葡萄糖衍生物的合成
称取 150 mg 上述合成的TpBD(NH2)2,1 g 干燥后的D-葡萄糖,20 mL 无水甲醇于 50 mL 的二颈烧瓶中,在 70 ℃ 条件下搅拌 4 d,待反应完成后,用纯水洗去未反应完的葡萄糖,烘干,得到红棕色的产物。合成路线如图2。

图2 TpBD(NH2)2的D-葡萄糖衍生物的合成路线
1.6 利用网包法制备TpBD(NH2)2的D-葡萄糖衍生物手性固定相
将TpBD(NH2)2的D-葡萄糖衍生物用玛瑙研钵研磨至比硅胶还细,称取 1.0 g 哌嗪于 50 mL 容量瓶内,加入异丙醇和 2 mL 三乙胺配制成 50 mL 溶液,称取 100 mg 研磨好的TpBD(NH2)2的D-葡萄糖衍生物加入到一定量配制好的上述溶液中,超声后浸泡上述硅胶3~ 4 h,吸出多余液体,在 110 ℃ 烘箱里加热处理 15 min,备用。
1.7 TpBD(NH2)2的D-葡萄糖衍生物手性固定相色谱柱的填充
称取 4.2 g 制备好的固定相,用有机膜滤过的正己烷/异丙醇(体积比9∶1)为溶剂,用250目的筛子,将固定相用溶剂湿法过筛,待固定相沉降后,倒出上清液。采用高压匀浆法装柱,在装柱机的溶剂罐中加入约 800 mL 的上述溶剂,向过筛后的固定相加入约 40 m L的上述溶剂,倒入匀浆罐前,为保证固定相分散均匀,要搅拌后立即倒入。拧紧装置后,保持 35 MPa 5 min,后将压力调为 25 MPa 保持 30 min,结束后打开减压阀,取下色谱柱,装上柱头,贴上标签标识上下端,装到液相色谱仪器上,用正己烷/异丙醇(体积比9∶1)为流动相,0.5 mL/min 的流速冲洗色谱柱,待基线稳定即可测样。
1.8 色谱拆分条件和性能评价参数
用正己烷/异丙醇(体积比9∶1;8∶2)和甲醇/水(体积比9∶1;8∶2)作为流动相,流速为 0.5 mL/min,紫外检测波长分别为 254、210,220 nm,柱温为 25 ℃。
对色谱柱的分离效果用保留因子k,分离因子α和分离度Rs进行评价。公式分别为:
k1=(t1-t0)/t0
(1)
k2=(t2-t0)/t0
(2)
(3)
(4)

2 结果与讨论
2.1 1,3,5-三甲醛间苯三酚(Tp)和3,3′-二硝基联苯胺(DNB)的表征
对1,3,5-三甲醛间苯三酚(Tp)进行核磁共振氢谱表征。1H NMR(500 MHz,CDCl3)δ:10.18(s,3H,CHO),14.15(s,3H,OH)。对DNB进行核磁共振表征。1H NMR(500MHz,DMSO-d6)δ:7.1(d,J=8.9Hz,2H),7.54(s,4H),7.74(dd,J=2.3Hz,2H),8.14(d,2H)。上述所得数据与文献报道[16]一致,表明已经成功合成Tp和DNB。
2.2 TpBD(NH2)2的D-葡萄糖衍生物手性硅胶球的表征
2.2.1 TpBD(NH2)2的D-葡萄糖衍生物的粉末X衍射表征分析
据文献报道,TpBD(NO2)2的PXRD图案在对应于(100)面的3.5°处显示出强烈衍射,6.0°、9.4°和26°的反射分别归因于(110)、(210)和(001)平面,属于AA堆叠模式。由图3看出,在3.5°、6.0°、9.4°、26°均出现了衍射峰,与文献报道一致[1-3],表明TpBD(NO2)2成功合成。经还原的TpBD(NO2)2和用D-葡萄糖修饰后的TpBD(NH2)2,其主要衍射峰也在3.5°、6.0°、9.4°、26°处,三者的峰形和出峰的位置基本相似,这也说明还原以及修饰后的物质的晶型和结构特征基本保持不变。

a.TpBD(NO2)2,b.TpBD(NH2)2,c.TpBD(NH2)2的D-葡萄糖衍生物
2.2.2 TpBD(NH2)2的D-葡萄糖衍生物的红外表征
TpBD(NO2)2、TpBD(NH2)2和TpBD(NH2)2的D-葡萄糖衍生物的红外表征如图4所示,从图4(a)TpBD(NO2)2的红外图可以看出,TpBD(NO2)2在 1512 cm-1和 1362 cm-1有吸收峰,对应于-NO2的特征吸收峰。从图4(b)TpBD(NH2)2的红外图显示,对比图4(a),在 1512 cm-1和 1362 cm-1处的峰强度减弱,说明TpBD(NO2)2中的-NO2被还原了。观察图4(c)在 1069 cm-1左右为C-O的特征吸收峰,对比图4(b),强度有所增强,说明D-葡萄糖成功修饰了TpBD(NH2)2。
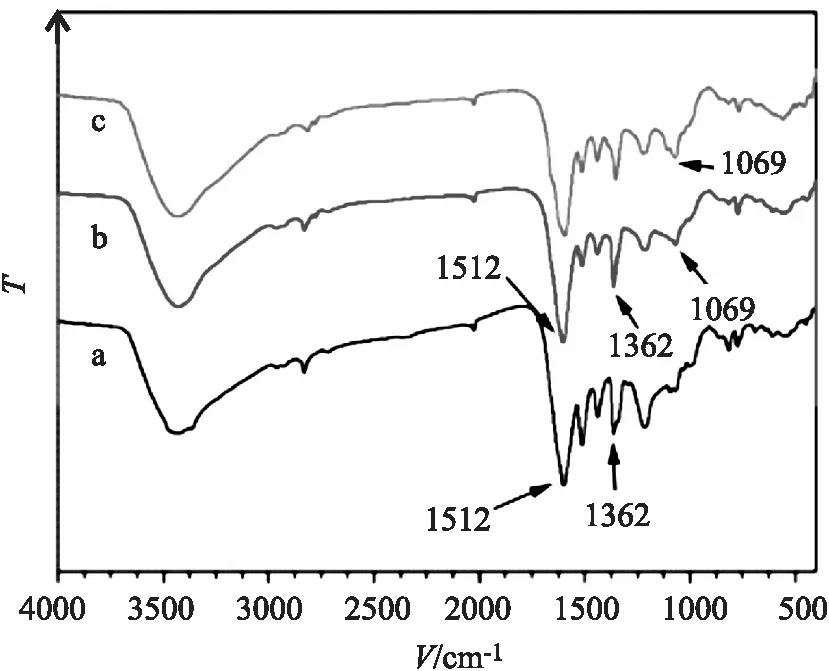
a.TpBD(NO2)2,b.TpBD(NH2)2,c.TpBD(NH2)2的D-葡萄糖衍生物
2.2.3 TpBD(NH2)2的D-葡萄糖衍生物氮气吸附测试
为了进一步了解合成的D-葡萄糖衍生物的结构特征,我们对其进行了氮气吸附测试,结果见图5。使用Brunauer-Emmett-Teller(BET)方法对测量数据进行计算,得到TpBD(NH2)2的D-葡萄糖衍生物的比表面积为 264 m2·g-1,孔体积为 1.78 cm3·g-1,通过Barrett-Joyner-Halenda(BHJ)分析计算测试数据,得到其平均孔径是 1.92 nm(图5b),符合相关文献报道的参数。

图5 TpBD(NH2)2的D-葡萄糖衍生物的氮气吸附测试
2.2.4 TpBD(NH2)2的D-葡萄糖衍生物的圆二色谱分析
如图6所示通过D-葡萄糖修饰后的 TpBD(NH2)2显示出了Cotton效应,表明葡萄糖成功修饰在COF上,使衍生后的TpBD(NH2)2具有手性。

图6 TpBD(NH2)2的D-葡萄糖衍生物圆二色谱图
2.2.5 TpBD(NH2)2的D-葡萄糖衍生物的扫描电镜
为了观察采用网包法后,TpBD(NH2)2的D-葡萄糖衍生物有没有固载到硅胶上,分别通过扫描电镜对空硅胶和网包后的固定相进行观察,结果见图7。通过电镜图片可以观察到,没有经过网包的硅胶表面光滑,没有任何物质固载在其表面,而经过网包的硅胶,其表面固载了大量的经过研磨的TpBD(NH2)2的D-葡萄糖衍生物,说明通过网包法,所合成的手性COF材料已成功固载在硅胶表面。

(a)纯硅胶,(b)TpBD(NH2)2的D-葡萄糖衍生物固定相
2.3 TpBD(NH2)2的D-葡萄糖衍生物手性固定相对于外消旋体的拆分
为了检测TpBD(NH2)2的D-葡萄糖衍生物制备的色谱柱的拆分能力,用其对外消旋体进行拆分。实验结果显示,所制备的手性色谱柱对15种手性物质能够进行不同程度的分离。其中CH3OH/H2O(体积比9∶1)的流动相对4种手性物质有一定的拆分效果,分别为二苯基乙醇酮、华法林、N-(3,5-二硝基苯甲酰)-L-亮氨酸、1-苯乙醇,而CH3(CH2)4CH3/(CH3)2CHOH的流动相对11种手性物质有一定的拆分效果,它们分别是2-甲基戊酸甲酯、2-乙基己酸、1,2-环氧丁烷、环氧溴丙烷、3-氯-2-丁酮、2-甲基戊醛、3-羟基丁酸乙酯、异丙基缩水甘油醚、乳酸乙酯、1-(1-萘基)乙胺、γ-辛内酯。它们的拆分结果见表1。

表1 色谱柱对15种手性物质的拆分结果
表1的分离结果显示,在CH3OH/H2O(体积比9∶1)的流动相条件下,所拆分的外消旋体中,二苯基乙醇酮和华法林做到了基线分离,拆分情况比较理想。在CH3(CH2)4CH3/(CH3)2CHOH的流动相条件下,2-乙基己酸、环氧溴丙烷、乳酸乙酯等达到了基线分离。两种流动相条件下,所拆分的物质不相同,能够相互补充,拓展了TpBD(NH2)2的D-葡萄糖衍生物手性固定相的拆分范围,该手性色谱柱所拆分的部分手性物质的谱图如图8所示。

图8 TpBD(NH2)2的D-葡萄糖衍生物色谱柱对外消旋体的部分图谱
测试结果显示TpBD(NH2)2的D-葡萄糖衍生物对一些手性物质具有比较好的分离效果,有些甚至能达到基线分离。能够对手性物质进行拆分的原因是,COFs材料上被修饰了D-葡萄糖手性基团,使COFs材料具备了手性,手性COFs材料对两种对映异构体存在保留差异,从而能够对手性化合物进行拆分。除此之外,手性固定相和手性物质之间的各种作用力,可能也在某种程度上对这些物质的分离作出贡献。
2.4 TpBD(NH2)2的D-葡萄糖衍生物液相色谱柱对于苯系位置异构体的拆分
为了探究TpBD(NH2)2的D-葡萄糖衍生物对于苯系位置异构体的分离情况,我们用所制备的色谱柱对苯系位置异构体进行了实验。在所分离开的这些物质中,该色谱柱对o,m,p-邻溴苯胺、o,m,p-硝基苯酚、o,m,p-氯苯酚能够进行一定程度的分离。它们的拆分结果见表2,拆分谱图见图9。

表2 色谱柱对2种位置异构体的拆分结果

图9 TpBD(NH2)2的D-葡萄糖衍生物液相色谱柱对苯系位置异构体的部分色谱拆分图
从图9中可以观察到,因苯系位置异构体的分子结构存在一些差异,因此导致它们在经过色谱柱时,该固定相对它们的作用不同,使得它们随着流动相从色谱柱中流出的时间不相同,即彼此的保留时间有差别,这也正是它们能够被拆分的原因。从分离图谱可知,TpBD(NH2)2的D-葡萄糖衍生物色谱柱对3种苯系位置异构体都有一定的拆分效果,这也拓展了TpBD(NH2)2的D-葡萄糖衍生物手性固定相在物质分离方面的应用。
2.5 温度对TpBD(NH2)2的D-葡萄糖衍生物色谱柱的影响
为了研究不同温度对TpBD(NH2)2的D-葡萄糖衍生物色谱柱出峰情况的影响,选用华法林进行测试。探究了25、30、35、40、45 ℃ 五个检测温度对TpBD(NH2)2的D-葡萄糖衍生物手性固定相出峰效果的影响。从对比谱图(图10)中可见,随着温度的升高,其保留时间在逐渐往后移动。因此可知,在一定温度范围内,随着温度的升高,对色谱柱的出峰情况没有太大影响,只有保留时间在后移,不影响出峰情况。

图10 不同温度下华法林在TpBD(NH2)2的D-葡萄糖衍生物在色谱柱上的分离谱图.
2.6 TpBD(NH2)2的D-葡萄糖衍生物色谱柱的重复性
为了探讨TpBD(NH2)2的D-葡萄糖衍生物所制备的色谱柱的重复性,选择华法林为代表,重复测样5次,其拆分谱图如图11所示,经过计算,第一个峰的峰面积与保留时间的相对标准偏差(RSD)分别为1.34%和1.25%。这一结果表明,TpBD(NH2)2的D-葡萄糖衍生物所制备的色谱柱具有良好的重复性。

图11 华法林在TpBD(NH2)2的D-葡萄糖衍生物色谱柱上的重复性分离谱图
3 结论
本文通过查阅相关文献制备了COFs结构 TpBD(NH2)2,使用后合成修饰的方法,让葡萄糖修饰到了该材料上,通过“网包法”固载到球型硅胶表面,获得了高效液相色谱手性固定相。用该固定相对二苯基乙醇酮、华法林和N-(3,5-二硝基苯甲酰)-L-亮氨酸等15种手性物质进行了分离。研究结果表明,本文合成的手性材料能够使多种手性物质实现不同程度的分离。其中CH3OH/H2O(体积比9∶1)的流动相对4种手性物质起到一定的拆分作用,CH3(CH2)4CH3/(CH3)2CHOH(体积比9∶1)的流动相对11种手性物质有一定的拆分效果。这些结果证明,采用后合成修饰的策略,将手性基团连接到无手性的共价有机框架上是可行的,手性的COFs材料用于液相色谱固定相具有一定的前景。
猜你喜欢
系统仿真技术(2022年4期)2023-01-17
分子催化(2022年1期)2022-11-02
云南化工(2021年8期)2021-12-21
世界农药(2019年3期)2019-09-10
中学生数理化·高二版(2016年3期)2016-12-26
国外医药(抗生素分册)(2016年4期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
信息记录材料(2016年4期)2016-03-11
合成化学(2015年10期)2016-01-17
华东师范大学学报(自然科学版)(2014年1期)2014-04-16
