
家族性噬血细胞性淋巴组织细胞增生症合并UNC13D基因突变3例病例总结并文献复习
2023-07-28 08:49:58钱冰涛王泽芯罗荣牡
罕少疾病杂志 2023年7期
钱冰涛 王 萌 王泽芯 罗荣牡 宋 庆
航天中心医院儿科 (北京 海淀 100049)
噬血细胞性淋巴组织细胞增生症(h e m o p h a go c y t i c lymphohistiocytosis,HLH)又称为噬血细胞综合征,可以累及任何年龄的个体,最常见于婴儿和年幼儿童,是一种危重的侵袭性免疫过度活化综合征。HLH临床表现上以持续性发热、肝脾肿大、全血细胞减少,多器官功能损害甚至衰竭,其病因是由于巨噬细胞、自然杀伤(NK)细胞、细胞毒T淋巴细胞等异常活化引起的全身过度炎症反应综合征[1]。本病罕见,临床表现不典型、复杂多样,且其病情进展迅速,病情危重,治疗难度大,死亡率高,诊断明确后应当及时治疗。近些年随着医疗技术发展以及对HLH的病因研究,各级医院临床医生对HLH的认识越来越深入,早发现,早诊断,早治疗,使得HLH临床诊治水平提高。
噬血细胞性淋巴组织细胞增生症(HLH)原发性和继发性两大类[2]。原发性HLH包括家族性HLH(FHL)及遗传性免疫缺陷综合征,由基因突变导致。目前为止已确认的FHL有5种类型:FHL1、FHL2、FHL3、FHL4、FHL5。其中4种FHL均已明确其致病基因,分别为PRF1、UNC13D、STX11、STXBP2。遗传性免疫缺陷综合征患儿往往具有免疫缺陷、生长发育异常等表现,包括Chediak-Higash综合征、Griscelli综合征、X连锁淋巴组织增生症、Wiskott-Aldrich综合征、严重的联合免疫缺陷病[3]。原发性HLH发病年龄早,多在婴幼儿期发病,故原发性HLH多发于儿童,成年人原发性HLH较为少见。
临床上最常见的是继发性HLH,可继发于感染性疾病、肿瘤、自身免疫性疾病、造血干细胞移植及药物过敏等。感染相关性HLH其病因感染源可以是病毒、细菌、真菌或寄生虫,临床上EB病毒感染导致的相关性噬血细胞综合征(EBV-HLH)最为常见[4]。儿童持续性EBV感染,可能会发展为慢性活动性EBV感染,从而导致继发性HLH[5-6]。
HLH在发病过程中存在巨噬细胞、NK细胞和CTLs持续活化,以至于产生过量细胞因子(细胞因子风暴),导致多脏器功能受损、多器官衰竭,这是HLH死亡率高的原因[7]。未经医院综合治疗的HLH自然病程几乎都是致命的。本病罕见且临床表现复杂,病因多样,故而及时识别HLH,并诊断明确HLH的病因,对于尽早行综合治疗改善预后至关重要。近些年基因检测技术飞速发展,HLH患儿诊断后进行基因检测,越来越多的家族性HLH(FHL)浮出水面,讨论原发性HLH患儿基因型不同,其临床表型是够有特异性。原发性HLH尽早评估病情,化疗后行造血干细胞移植(hematopoietic cell transplant, HCT),有助于改善患儿预后。本研究对3例家族性HLH(FHL)临床资料进行了收集,分析患儿的临床特点及实验室、功能学检查结果的差异,为HLH患儿的个体化治疗提供一定依据。
1 资料与方法
1.1 一般资料资料选取2020年5月至2022年10月北京航天中心医院儿科住院并确诊为家族性HLH(FHL)临床资料进行回顾性分析,均通过航天中心医院医学伦理委员会批准,且监护人签署知情同意书。随访:3例FHL患儿来院门诊复查或住院治疗进行随访。
纳入标准:3例均符合中国专家共识(2018年版)中噬血细胞综合征诊断标准[8]。(2)3例患儿均行噬血细胞综合征相关基因检测。HLH累及中枢神经系统(CNS-HLH)诊断标准国际目前没有统一定论[9]。很多专家认为中枢神经系统受累指的是HLH患者出现一种或多种中枢神经系统和/或精神症状、神经影像学、脑脊液的异常,并且排除其他因素。CNS-HLH可以分为三个病理学阶段:Ⅰ期:活化的淋巴细胞、巨噬细胞只浸润软脑膜;Ⅱ期:浸润周围血管间隙;Ⅲ期:脑实质的广泛浸润,引起血管破坏及脑组织的广泛多灶性坏死、脱髓鞘病变,最后出现神经元丢失和胶质增生[2]。
1.2 方法回顾性分析3例家族性噬血细胞性淋巴组织细胞增生症(FHL)患儿的临床资料,从病历中收集并分析患儿的临床表现,常规实验室检查结果,EB病毒抗体及血清EBV-DNA检测结果,T、B、NK细胞检测结果,影像学检查报告,遗传学测序结果(原发性HLH相关基因筛查)以及自然杀伤(NK)细胞活性、可溶性CD25分子(sCD25正常值<6400ng/L)。
2 结 果
2.1 一般资料收集3例家族性噬血细胞综合征FHL,3例患儿UNC13D基因突变均阳性,均行化疗后进行造血干细胞移植(hematopoietic cell transplant, HCT)。
2.2 临床症状及体征病例1,5岁,女性,以“皮疹、发热、淋巴结肿大”起病,病初头面部、全身出现丘疹样皮疹,高出皮面,部分破溃、渗出,间断发热,体温最高达39℃,热峰1次日,流黄涕,鼻孔粘膜增生,查体耳后、颈部淋巴结肿大,腹部触诊肝脾肿大。皮疹好转后,患儿发热、抽搐1次,查头颅核磁MRI提示胼胝体压部有异常信号,考虑伴胼胝体压部存在可逆性病变、临床症状轻微的脑炎脑病(Mers)。小脑脑沟稍宽。左顶头皮下异常信号,考虑皮样囊肿可能。EB病毒抗体检测:EB病毒衣壳抗原(CA)IgG阳性;EB病毒核抗原(NA)抗体阳性。淋巴结病理示:右颌下淋巴结EB病毒阳性T细胞增殖性疾病(Ⅱ级)。复查头颅核磁提示脑室系统稍扩大,脑沟脑裂略增宽,轻度脑白质发育异常,轻度鼻窦炎。给予化疗及对症治疗,并行造血干细胞移植术,定期随访。

表1 噬血细胞综合征临床特点
病例2,3岁,男性,以“腹痛、腹胀”起病,之后出现发热,查体颈部淋巴结肿大,肝脾肿大,完善骨髓穿刺术送检未见噬血细胞现象,EB病毒DNA测定1.09×104copies/mL,抗病毒治疗无效,患儿间断发热,出现皮疹,该患儿病史、体征以及实验室检查,临床诊断HLH,复查骨穿可见噬血细胞现象,诊断噬血细胞性淋巴组织细胞增生症(HLH),给予化疗及对症治疗,并行造血干细胞移植术,定期随访。
病例3,6月,男性,以“发热、皮疹”起病,病初高热,体温39℃,热峰4次/日,散在红色皮疹,抗感染治疗效果不佳,持续高热,根据患儿临床表现、体征以及实验室检查诊断噬血细胞性淋巴组织细胞增生症(HLH),骨髓穿刺术送检骨髓可见噬血细胞现象,给予化疗及对症治疗,并行造血干细胞移植术,定期随访,生长发育落后。
2.3 临床实验室检查资料整理
2.4 影像学检查资料
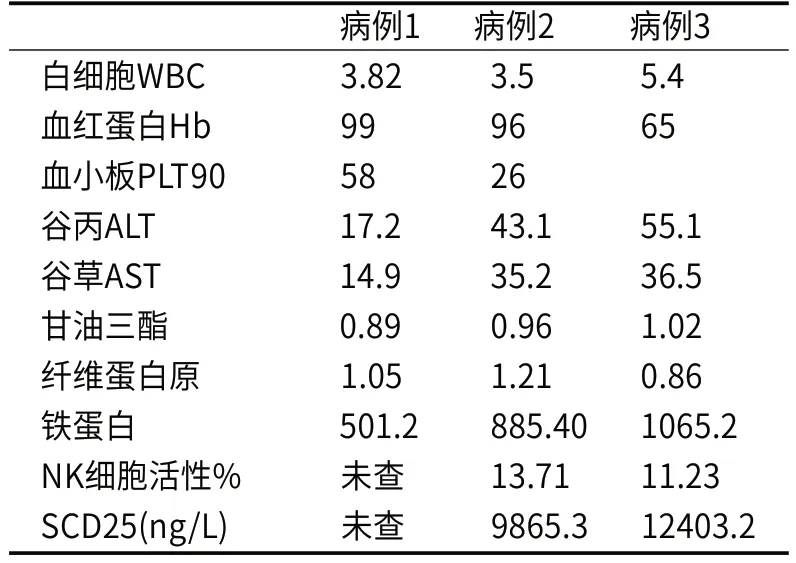
表2 临床实验室检查结果
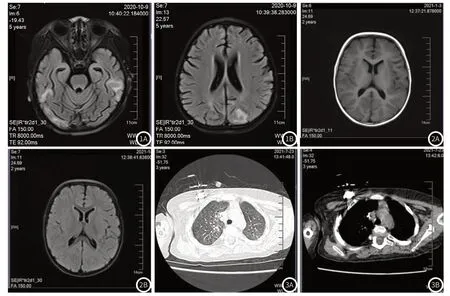
病例1 5岁,有发热、抽搐症状,头颅核磁提示双侧侧脑室周围白质内可见晕状T1WI等信号、T2WI高信号、FLAIR高信号灶。DWI未见异常高信号。脑室系统稍扩大,脑沟脑裂略增宽。轻度脑白质发育异常。(见图1A-图1B)病例2 6月发病,生长发育落后,头颅核磁:左侧放射冠可见点状T2WI及FLAIR高信号,左侧放射冠点状异常信号灶,脱髓鞘。DWI未见明显高信号。左侧颞部蛛网膜囊肿。(见图2A-图2B)病例3 6月发病,反复感染性肺炎,出现并发症闭塞性支气管炎,胸部CT提示支气管管壁增厚,双肺多发淡片状磨玻璃影,双肺下叶呈马赛克改变,右肺尖胸膜下索条影,右肺中叶、左肺下舌段条片状实变影。(见图3A-图3B)
3 讨 论
噬血细胞性淋巴组织细胞增生症(HLH)发病机制中涉及的细胞类型包括2类:1.巨噬细胞是起源于循环中单核细胞的专职抗原提呈细胞,可将外源性抗原提呈给淋巴细胞。HLH巨噬细胞出现异常活化,并分泌过量的细胞因子,最终导致组织重度损伤,可引起器官功能衰竭。2.自然杀伤细胞(natural killer cell)占到淋巴细胞数的10%-15%。NK细胞通常可以清除受损或被感染的宿主细胞(例如巨噬细胞)。HLH患儿体内NK细胞和/或CTLs不能够清除那些异常活化的巨噬细胞。由于缺失以上正常反馈调节,CD8+ T细胞和巨噬细胞出现过度活化,IFN-γ、细胞因子水平异常升高,从而导致HLH的病理变化[10-11]。HLH患儿体内免疫系统Toll样受体(tolllike receptor, TLR)活化可能是导致HLH的另一个原因[12]。
HLH其他免疫紊乱,比如淋巴细胞异常包括CD4和CD8淋巴细胞亚群数量改变。2015年纳入21例HLH患者中有20例出现淋巴细胞亚群和/或免疫表型异常。其中10例出现CD8+细胞增多和CD4:CD8比率降低,8例出现CD3+细胞减少,3例出现CD56+细胞增多、CD7-/CD4+细胞增多和CD4+细胞增多。病例系列研究显示:CD8数量增加和CD4/CD8比值下降的患者生存情况最佳。CD3总数下降预示着患者预后较差[13]。
收集到的3例原发性家族性HLH均为UNC13D基因突变阳性。在原发性HLH携带破坏性基因突变的患儿发病年龄较早;家族性HLH(FHL)的患儿较继发性HLH更容易出现中枢神经系统受累,出现抽搐、嗜睡、精神异常等;FHL脱颗粒通路受损患儿sCD25、胆红素及IFN-γ、IL-10水平较高[14]。Unc(即uncoordinated)蛋白家族调节溶细胞颗粒的成熟,UNC13D/Munc13-4-编码Munc13-4的UNC13D基因突变会导致FHL3[15]。
HLH除了单个HLH基因的纯合性突变外,HLH也可能为复合杂合子突变(同一基因的两个等位基因上可能存在不同的突变)或为双基因遗传(在两个不同的基因上可能存在不同的突变)。一项回顾性研究纳入了2701例HLH就诊接受基因检测的患者,结果发现225例(8%)患者为纯合性突变或复合杂合型突变,其中28例(1%)患者为双基因遗传,有21例在PRF1和脱颗粒基因内发生突变,7例在参与脱颗粒途径的2个基因内发生突变。此项大样本研究表明,由于参与细胞毒性淋巴细胞脱颗粒的基因内的协同功能效应,FHL具有潜在的双基因遗传模式[16]。
另一项研究报道了类似结果,纳入了281例归为“散发性”HLH的患者中,43例存在已知的家族性HLH基因的单等位基因突变,提示该病并非单纯通过隐性遗传。基因SPRF1(FHL2)和UNC13D(FHL3)的突变占FHL病例的70%。因此90%以上的家族性HLH(FHL)患者可以进行基因诊断。穿孔素表达和脱颗粒程度对FHL的诊断比噬血细胞和细胞毒性试验更有用。FHL并不是严格的隐性遗传。专家们认为临床综合征HLH通常是由外源性触发因素和遗传易感性共同作用的结果。外源和遗传因素的不同权重解释了从HLH继发到严重感染到FHL的广泛疾病谱[17]。并且尽早进行基因检查,有助于明确是否为FHL,有助于制定治疗计划,尽早行骨髓移植治疗。
一篇文章报道了1例患儿,主要表现为发热,伴腹胀、鼻衄,当地医院诊断慢性活动性EB病毒感染,伴有肝功能异常、肝脏纤维化,入院后诊断为继发性HLH,住院对症治疗好转后出院,1月后随访患儿病情恶化,因多脏器功能衰竭死亡[18]。因此本病容易出现多脏器受累,病情进展迅速,对症治疗及化疗治疗效果不理想,预后极差。家族性HLH(FHL)携带基因突变,病情更为凶险,应尽早完善相关检查评估病情,尽早行全面治疗。
综上所述,家族性HLH(FHL)发病年龄较小的患儿遗传信息有助于评估其HLH复发风险、造血干细胞移植的必要性,以及家庭成员出现HLH的风险。HLH发病年龄越小,发现基因突变概率越高,因此对于小年龄起病的反复发热、皮疹、肝脏脾脏肿大患儿,诊断为噬血细胞综合征建议尽早进行基因突变的筛查。基因筛查有异常,确诊为家族性HLH(FHL)病情进展迅速,化疗及对症治疗后,建议尽早行造血干细胞移植有助于改善预后。
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
中国生殖健康(2020年2期)2021-01-18 02:51:26
河南科学(2020年3期)2020-06-02 08:30:16
云南医药(2019年3期)2019-07-25 07:25:16
铜仁学院学报(2018年6期)2018-07-05 09:47:36
小学生导刊(2018年13期)2018-06-29 03:49:00
哈尔滨医药(2016年1期)2017-01-15 13:43:18
中国继续医学教育(2015年3期)2016-01-06 01:36:46
中国药业(2014年4期)2014-05-09 08:48:33
西部中医药(2014年6期)2014-03-11 16:07:43
