
蛋白质三维结构解析的方法
2023-07-13 02:28徐甲强崔倩倩
河南师范大学学报(自然科学版) 2023年4期
徐甲强,崔倩倩,2
(1.上海大学 理学院化学系;新能源与传感技术实验室,上海 200444;2.中国科学院 上海药物研究所,上海 201203)
蛋白质是执行生命活动过程中重要的生物大分子,参与肌肉收缩、能量转换、氧气输送以及抵御微生物入侵等各种生理过程.虽然蛋白质的功能各不相同,但是其基本组成是类似的.其通过20种氨基酸排列组合形成不同的肽链,再进一步地组装和折叠,成为具有独特且复杂三维结构的生物大分子[1].结构的正确解析是理解蛋白质生物学功能、认识各蛋白质之间相互作用的关键[2],可以为药物的研发提供重要的靶点信息[3].
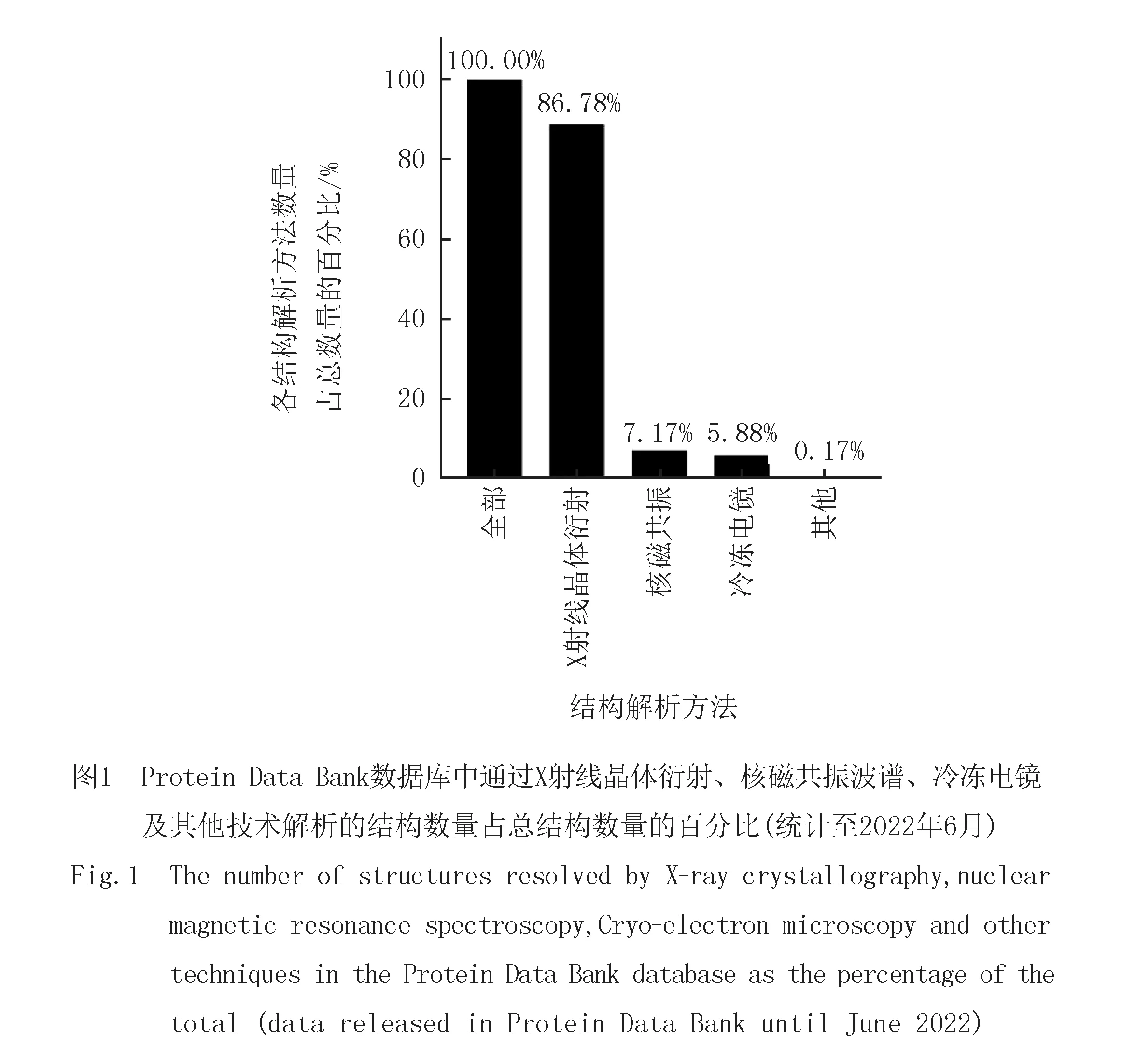
目前蛋白质结构解析的方法分为实验方法和预测方法两类,前者主要包括X射线晶体衍射(X-ray Crystalline Diffraction)法、核磁共振(Nuclear Magnetic Resonance,NMR)法和冷冻电子显微镜(Cryogenic Electron Microscopy,Cryo-EM)法,这3种方法逐渐发展成熟,已成为蛋白质结构实验分析的三大技术平台.后者主要以AlphaFold人工智能系统的建立为标志,基于大数据的计算对蛋白质结构进行预测.各种结构解析方法占比情况见图1.
1 X射线晶体衍射法
生物大分子结构测定技术的历史可以追溯到19世纪末.1895年,德国物理学家伦琴发现了X射线.X射线是一种高能量、短波长的电磁波(本质上属于光子束),当X射线击打在分子晶体颗粒上时,因与晶体中的电子发生作用而产生衍射效应.通过探测器收集这些衍射信号,可以确定晶体中电子密度的分布,从而获得粒子的位置信息,为解析生物大分子的晶体结构提供依据.X射线衍射法凭借其优异的性能在接下来的一个多世纪获得了极大发展,被广泛用于生物医学和工业领域.只要是能够形成结晶的蛋白质,通过X射线衍射后就能看清楚其三维结构,因此X射线晶体衍射法成为解析蛋白质大分子晶体结构的主要手段,并逐渐发展为一门学科即X射线晶体学(X-ray Crystallography).X射线晶体学成为最早且最主要研究测定蛋白质结构的学科[4].
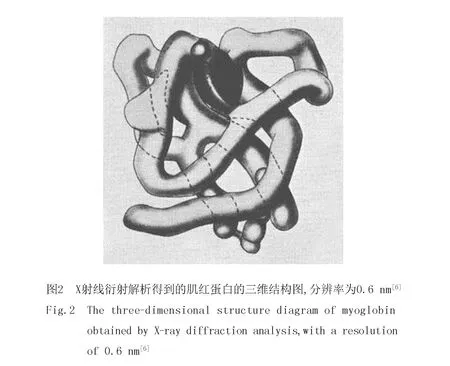
1934年,BEMAL和CROWFOOT获得了第一张蛋白质晶体的X射线衍射照片,但是由于理论的局限性,并未解析出蛋白质的三维结构[5].1958年,英国科学家MAX PERUTZ和JOHN KENDREW将重金属原子引入蛋白质晶体中,通过运用多对同晶置换法(Multiple Isomorphous Replacement,MIR)成功解决蛋白质大分子晶体衍射的相位问题,并得到了高分辨率的肌红蛋白(Myoglobin)的三维结构[6](如图2),以及更复杂的血红蛋白(Hemoglobin)的晶体结构[7].自此,X射线衍射技术在蛋白质结构测定中得到了实际的应用,PERUTZ与KENDREW也获得了1962年的诺贝尔化学奖.1964年,AARON KLUG将X射线晶体学与电子显微镜结合起来,发明了一种可以解析更大的蛋白质或蛋白质-核酸复合物结构的新的分析仪器——电子晶体学显微镜(Crystallographic Electron Microscopy)[8].由于KLUG在结构生物学领域的杰出贡献,被授予了1982年的诺贝尔化学奖.1965年,BLAKE和PHILLIPS用X射线衍射技术测定了溶菌酶的三维结构并首次解释了蛋白质的分子机制[9-10].但由于此时产生的X射线存在相应的缺陷,如没有相干性、亮度不高、偏振性较差等,在很大程度上限制X射线的应用,直到同步辐射技术的出现才使得部分难题得到解决.1947年,学者在美国通用电气公司的一台70 MeV的同步加速器中首次观察到“同步光”现象,并将该现象命名为同步辐射.同步辐射是指速度接近光速的带电粒子在做曲线运动时,沿切线方向发出的电磁辐射.该辐射拥有高强度、宽波谱、高准直性及方便可调节的入射X射线等优点,弥补了之前X射线在亮度和偏振性方面的不足,使得X射线晶体学得到了极大的发展[11].1976年,KEITH HODGSON首次使用同步辐射装置对单个蛋白质晶体进行照射,观察到生物大分子的电子和结构环境[12].然而,蛋白质结晶过程并不简单,有时需要改变样品的结构才能得到有序的晶体,这使得人们可能会错过蛋白天然状态下的结构信息,因此科学家们尝试在非结晶的状态下解析蛋白质结构.同步辐射的发展为小角X射线散射(Small Angle X-ray Scattering,SAXS)提供了高强度的入射光源,实现对样品的快速曝光,因此,与同步辐射只能解析静态或微动态的蛋白质结构不同,应用SAXS技术可以获得溶液状态下蛋白质结构的动态变化.然而,科学家们对X射线晶体学的探索不止于此.21世纪初,德国率先建造了X 射线自由电子激光器(X-ray Free-Electron Laser,XFEL),实现了对生物大分子皮秒(ps)量级甚至飞秒(fs)量级的动态结构的观察[11].大分子结构的预测不再局限于静止状态,已经进入了时间分辨晶体学的新时代.
自20世纪90年代以来,X射线晶体学迎来了飞速发展的时期.直至今天,X射线晶体衍射技术仍是最常用且最准确的测定蛋白质三维结构的技术[13].目前PDB(Protein Data Bank)数据库中已经收录了超过18万个生物大分子结构,如图1所示,其中约86.78%是通过X射线衍射技术获得的.X射线晶体衍射技术虽然得到了长足的发展,但仍存在一定的缺点.比如,X射线本身带有极大的能量,容易对晶体造成辐射损伤,即使是液体状态下的蛋白质也会受到影响.此外,X射线衍射方法不能用来解析分子量较大的蛋白质,且对于不能结晶的蛋白质的解析也存在一定的局限性.随后核磁共振波谱学的发展在一定程度上弥补了X射线晶体衍射技术的不足,逐渐发展成为蛋白质结构解析的新方法.
2 核磁共振波谱法
利用核磁共振波谱进行的结构研究可以观察到蛋白质分子的天然结构和动力学特征[14].核磁共振的基本理论是带有孤对电子的原子核在外界磁场的影响下发生自旋,并通过塞曼分裂发生能级跃迁,从而吸收并释放电磁辐射,产生共振频谱.这种共振电磁辐射的频率与所处磁场强度成一定比例.由于不同分子中原子核所处的化学环境不同,将会产生不同的共振频率,从而产生不同的共振谱.通过记录波谱的变化可以判断该原子在分子中所处的位置及相对数目,用以进行定量分析及分子量的测定,并对分子进行结构分析.
1938年,美国科学家ISIDOR ISAAC RABI首次通过分子束实验发现在磁场中的原子核会沿磁场方向呈正向或反向有序且平行的排列,在施加外磁场后,原子核的自旋方向发生翻转[15].这是人类关于原子核与磁场以及外加射频场相互作用的最早认识.1946年,PURCELL和BLOCH都独立观察到了固体(石蜡)和液体(水)状态下的核磁共振吸收[16-17],两位科学家也因此获得了1952年的诺贝尔物理学奖.1948年,核磁弛豫理论建立.1950年,化学位移和自旋耦合现象被相继发现.之后,RICHARD R ERNST团队于1966年建立了傅立叶变换核磁共振分光法,并于1976年建立二维核磁共振技术.这两项技术极大地提高了NMR测量的灵敏度和分辨率,并将核磁共振成功应用到化学、结构生物学和日常的医学诊断中[18].随后,WUTHRICH将该方法成功应用于生物大分子结构与动力学研究[19].这些理论学说的建立使得NMR快速发展.1978年,核磁共振技术首次被用于蛋白质结构的解析[20].经过半个多世纪的发展,尤其是高场核磁共振谱仪的出现、各种新的核磁实验方法的应用、分子生物学的进步以及各种标记方法(如同位素标记技术)的产生等使得核磁共振技术在结构生物学中得到广泛应用[18].直至今日,核磁共振波谱技术仍然是能够在原子分辨率下测定溶液中生物大分子三维结构的最可靠的方法之一.如图1所示,PDB数据库中收录的生物大分子三维结构中约有7.17%是通过NMR技术获得的.
核磁共振法解析蛋白质结构的一般流程如下:1)对蛋白质进行同位素标记,通常用15NH4Cl为氮源、13C-葡萄糖为碳源的M9培养基培养大肠杆菌并表达蛋白[21](现在逐渐发展成为对氨基酸的选择性标记);2)将蛋白放置到强磁场中,用射频信号进行探测,进行1H、13C、15N三共振NMR实验,观测到的共振信号可以反映相邻原子核之间的相互作用和成键原子之间的局部构象,即对蛋白质的主、侧链进行归属[22-23];3)对采集完的NMR谱图进行氨基酸的比对,结合NOE(核欧沃豪斯效应增强)约束、二面角约束(通过耦合常数所得)以及空间距离约束(由三维15N-13C-NOESY-HSQC实验获得),来构建蛋白质原子模型;4)结合计算机不断演算,得到最终的蛋白质结构.图3为文献[24]中利用核磁共振光谱测定溶液状态下的锌指核酸结合基序的三维结构图.
NMR结构解析大多是对溶液状态下的蛋白质结构进行非破坏的检测.因此一般认为比起X射线晶体衍射,NMR能更准确地描述生物大分子的天然结构.核磁共振波谱技术适合研究瞬时存在和不稳定的复合物.如采用半滤波的核欧沃豪斯效应谱(Half-Filrered NOESY)可以测定存在弱相互作用复合物的三维结构[25-26].同时,核磁共振技术也适合研究蛋白质动力学,其时间尺度可以囊括皮秒到秒的范围[18],可精确得到目的基团的动态变化信息.此外,利用液体或固体核磁共振的方法,逐步突破了分子大小以及长程约束的限制,使得NMR还可以研究膜蛋白的三维结构,特别是一些通过X射线晶体衍射技术和Cryo-EM技术不能确认的蛋白质的柔性区域的结构信息.当然,NMR也存在一些劣势,如蛋白质在溶液中存在结构不稳定的情况,这种状态下很难获取稳定的NMR信号.此外,由于大分子量的蛋白质在NMR谱中存在重叠峰的问题,因此NMR技术在处理大型生物分子时的能力有限[14].而冷冻电子显微镜技术突破了蛋白质分子量的限制,成功打破X射线晶体衍射技术和NMR技术的壁垒,成为当前结构生物学领域最前沿的成像技术之一,极大地推动了膜蛋白及大分子复合物的结构解析进程.
3 冷冻电子显微镜法
使用冷冻电子显微镜对液体样品进行玻璃化处理,集中了X射线晶体衍射和核磁共振波谱两种方法的优点,成为结构研究最合适的替代方法[14].冷冻电子显微镜技术的原理是将生物大分子在毫秒的时间尺度内快速冷冻并包埋在玻璃态的冰中,应用低温透射电子显微镜收集样品信息,即用高度相干的电子做光源,采用80~300 kV加速电压将电子束加速,透过样品和附近冰层,使样品受到散射,再利用探测器和透镜系统把散射信号记录下来,收集生物大分子不同方向上的二维投影,再经图像处理并利用三维重构的方法计算得到大分子三维精细结构.
电子显微镜最早出现在1931年,其设计之初的目的就是为了获得高分辨率的病毒图像.但由于电子显微镜必须在高真空下工作,而生物样品大多在真空状态下不能保持稳定甚至失活,因此制备高分辨率的生物样品是当时制约电子显微镜技术发展的主要瓶颈,导致其在结构生物学方面的应用要远远落后于材料科学.在冷冻电镜技术发展之前,一般使用重金属盐覆盖生物样品,用常温负染透射电镜进行观察,但是获得的生物大分子分辨率较低[27].
1968年,DE ROSIER和KLUG成功地用负染的电子显微镜照片重建了噬菌体尾部(一种螺旋对称的粒子)的三维结构,开创了电子显微镜三维重构技术,取得了蛋白质结构解析的重大突破[28](图4).1974年,ROBERT GLAESER和KEN TAYLOR首次提出对含水生物样品冷冻后再进行电子显微镜成像,通过测试发现该方法可以有效降低辐射损伤对样品高分辨率结构的破坏,开创了维持高真空状态下高分辨率成像的新思路,这就是冷冻电子显微镜的雏形[29].1975年, HENDERSON等人开创了二维电子晶体学三维重构技术,即应用电子显微镜从二维晶体中获得近原子分辨率的三维蛋白质结构,并使用该方法解析了第一个膜蛋白的三维结构——视紫红质蛋白[30].FRANK等人建立了基于冷冻电镜单颗粒重构技术的图像分析方法,能够从2D电子显微图像构建3D结构[31].DUBOCHET教授创建了样品制备的低温玻璃化法,该方法通过使用液氮冷却的液态乙烷实现了毫秒之内的生物样品的快速冷冻,成功制备了不形成冰晶的玻璃态冰包埋的生物样品,能够使生物分子在真空中保持其自然状态,并有效抵御电子的辐照损伤[32].上述开拓性的科研成果共同奠定了冷冻电子显微镜技术发展的基础,并获得了科学界的认可.HENDERSON、FRANK和DUBOCHET 3位科学家获得了2017年的诺贝尔化学奖.

起初,使用冷冻电子显微镜技术只能解析高对称性且大分子量的病毒颗粒的近原子分辨率的三维结构[33],而后这项技术取得了革命性的进步,其主要的原因之一是电子直接探测器(Direct Electron-Detector Device,DDD)的发展.DDD允许完全相同的图像区域的多个图片通过帧对齐和取平均的方法校正光束引起的背景运动,显著提高图像的信噪比(Signal-to-Noise Ratio,SNR)[14].除了图像处理硬件的突破之外,冷冻电子显微镜技术的进步还得益于图像处理软件的发展[34].2012 年SJORS SCHERES开发的RELION算法能更有效地处理低信噪比的图像,这成为单颗粒结构解析的利器.基于前人的研究基础,2013年,新一代DDD相机(Gatan K2 camera)拍摄了近九万张单颗粒图像,并解析得到了约300 kDa的膜蛋白——瞬时受体电位通道蛋白(TRPV1)四聚体的冷冻电镜结构,分辨率高达近原子级别的0.34 nm[35],标志着冷冻电子显微镜技术进入“原子分辨率”时代,并掀起了一场 “分辨率革命”.
近年来,通过冷冻电子显微镜测定的大分子复合物的高分辨率结构的数量呈现指数级的增长趋势,同时结构解析可达到的最高分辨率也在不断提高.2014年,英国SCHERES团队通过改进电子显微镜技术,成功获得了酵母菌的线粒体核糖体大亚基的图像,分辨率达到0.34 nm[36].2015年,清华大学施一公教授团队首次在世界上揭示了分辨率高达0.43 nm的人体γ-分泌酶的电镜结构[37],这为理解γ-分泌酶的工作机制及阿尔茨海默病的发病机理提供了重要基础.2016年,美国国家癌症研究所科学家发布的谷氨酸脱氢酶(分子量334 kDa)结构的分辨率已经达到了0.18 nm[38].2020年,德国HOLGER STAR课题组解析了脱铁蛋白(Apoferritin)的三维结构,并达到了原子级别的分辨率(0.125 nm),直接可视化蛋白质中的所有原子(包括氢原子)[39],打破了之前日本KEIICHI NAMBA团队保持的0.154 nm的记录,实现了世界首次可视化蛋白质中的单个原子.相信随着冷冻电子显微镜技术的发展,这个纪录将会被不断打破.
综上所述,突破分辨率限制的生物分子一般有着分子量大、化学性质稳定、结构对称性好等特点,而对于小分子量蛋白质(一般为小于200 kDa)颗粒由于其在冷冻样品中衬度不足等原因,其高分辨解析工作对目前的技术手段而言仍然是很大的挑战.但是这项技术壁垒随着冷冻电子显微镜单颗粒分析技术(Single Particle Analysis,SPA)的发展逐渐被打破.2019年,FAN等[40]解析的分子量为52 kDa的链霉亲和素蛋白的分辨率达到0.32 nm.该研究成果一度刷新利用冷冻电子显微镜单颗粒方法解析至近原子分辨率生物大分子分子量最小记录.2022年3月,斯坦福大学医学院ZHANG等[41]通过设计蛋白质双壳来克服尺寸限制,将环磷腺苷效应元件结合蛋白(CAMP-response element binding protein,CREB)中结合蛋白(CREB binding protein,CBP)的KIX结构域(11 kDa)夹在Apoferritin内壳和麦芽糖结合蛋白外壳的中间,构成分子笼,并获得了分辨率为0.26 nm的三维结构.这项研究表明,将小分子量蛋白质融合为笼状结构是打破冷冻电子显微镜结构测定的分辨率极限的可行方案,但是该方法的适用性仍需要通过实验进一步验证.
随着时间的推移,冷冻电子显微镜技术已经向4个方向上扩展,即电子二维晶体学(Electron 2D Crystallography)[42]、单颗粒分析(SPA)[43]、冷冻电子断层扫描(Cryo-Electron Tomography,Cryo-ET)[44]以及最新的微晶电子衍射(Microcrystal Electron Diffraction,MicroED)[45].目前占据主导位置的是单颗粒分析技术,也是推动“分辨率革命”的主要方法.通过使用重组颗粒方法,例如洗涤剂胶束[46]或纳米圆盘[47],单颗粒冷冻电镜已经能够克服旧方法在结构异质性方面的困难[48].而随着近些年聚焦离子束(Focused Ion Beam,FIB)减薄技术和相位板[49]等技术的发展,以及光镜-电镜结合实现样品定位策略的应用,使得更大尺度上的冷冻电子断层成像技术逐渐发展成熟.Cryo-ET的适用尺度非常广泛,包括从分子水平的蛋白质到亚细胞水平的细胞器,以至细胞水平的组织结构,都可以被观察到,这项技术也已经被成功应用于直接观察病理条件下细胞及组织内部的目标大分子复合物的定位、互作及引发疾病的构象变化机制.Cryo-ET有效填补了X射线晶体学、核磁共振以及SPA等方法得到的高精度结构和光学显微镜技术得到的低分辨率的细胞整体图像之间的空白,已经成为连接结构生物学和细胞生物学的重要桥梁[27].
冷冻电子显微镜解析蛋白质三维结构方法的优点是可直接获得生物大分子在接近生理条件下的结构及构象变化、无需结晶、所需样品量相对较少、研究对象广泛,适合解析大分子复合物(分子量大于80 kDa的蛋白质、病毒及复合物)的三维精细结构,且样品分子量越大、对称性越高,越容易区分异质性,从而越容易获得高分辨率结构[33].此外,作为当前结构生物学领域最前沿的成像技术之一,冷冻电镜技术已经将生物大分子复合物的结构解析能力拓展至原子分辨率水平.“分辨率革命”使Cryo-EM成为结构引导药物设计领域的未来工具[50-52],在膜蛋白结构解析和小分子药物及疫苗的开发中显露出巨大的潜力[27].
尽管仍然面临一些挑战,如冷冻电子显微镜对于样品制备的要求较高,特别是分子量较小、均一性差的样品,获得的分辨率较低,设备及实验成本昂贵等,但是科技的突破还在不断进行,冷冻电子显微镜技术未来可期[14].
4 AlphaFold人工智能预测蛋白质结构
由于上述方法实验测定过程十分复杂,且成本高、费时,对某些不易结晶的蛋白质还存在局限,因此,随着计算机技术的发展,以生物信息学为基础发展起来的蛋白质结构预测方法逐渐受到重视.
蛋白质结构预测的方法主要分为两类,第一类为理论分析方法,即通过理论计算进行结构预测,该类方法假设折叠后的蛋白质取能量最低的构象,理论上是可行的,但是从实际出发,该方法存在缺陷:1)天然状态与未折叠状态下的蛋白质结构之间的能垒很小,结构变化细微;2)蛋白质可能的空间构象数量庞大,针对蛋白质折叠方式的计算量巨大;3)计算模型中参数的不确定性,这些缺点限制了理论分析方法的发展.第二类为统计方法,实质上是根据蛋白质的编码基因推测出可能的氨基酸序列,之后对已知蛋白质结构进行统计分析,建立序列到结构的映射模型,根据已掌握的蛋白质的结构特点推测未知蛋白质的可能结构,并以此结构为基础,预测该蛋白质的生物学功能[2].目前蛋白质结构预测的方法主要以第二类为主.
20世纪70年代,美国科学家ANFINSEN提出蛋白质的氨基酸序列决定其三维结构的概念,成为开展蛋白质结构预测的基本信念[53].也正是由此开始,计算科学家开始进入蛋白质结构预测领域,并建立计算机模型,以预测给定的蛋白质如何折叠.起初建立的模型只适用于小分子量蛋白质或大分子量蛋白质的短片段,此外,由于前期实验方法解析蛋白质结构的进度缓慢,无法提供足够多的可供参考的蛋白质三维结构模型,导致基于算法的蛋白质结构预测也是进展缓慢.直到20世纪90年代,X射线晶体学、冷冻电子显微镜等技术的突飞猛进使得PDB数据库中的蛋白质三维结构数量与日俱增,极大推动了计算生物学的发展.1994年,美国科学家MOULT和他的同事共同发起了两年一次的国际蛋白质结构预测竞赛(CASP).组织方提供大约一百种已经通过实验方法解析出三维结构但未向公众发布的蛋白质的氨基酸序列,参赛者通过算法预测蛋白质结构,之后将预测结果与实验结果进行比较并评分(Global Distance Test,GDT),只有大于90分才被认为该算法与实验方法相比有竞争力[54].此后的二十多年间,竞赛中出现的最高的计算机测试成绩也只达到了40分,直到2018年,谷歌公司旗下的DeepMind公司研发出的AlphaFold首次参加CASP,并获得了历史最高的近80分的好成绩,但是预测准确度仍有一定差距.时隔两年,在2020年的第14届CASP中,DeepMind团队拿出升级版的AlphaFold2[55]系统,竞赛评分达到了92.4分,在蛋白质结构预测方面表现出了无与伦比的准确性,甚至可以分析X射线晶体学难以解决的存在跨膜结构域的蛋白质结构.AlphaFold2系统优越的表现引起了科学界的热切关注.
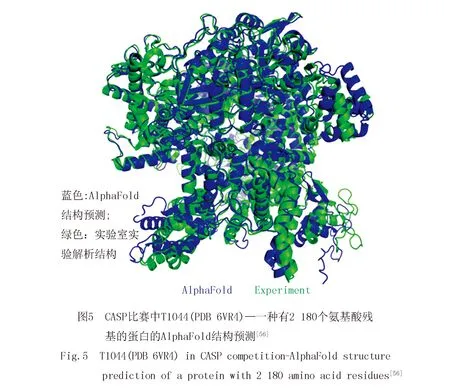
2021年7月,DeepMind团队在Nature杂志上连续发文,首先公布了AlphaFold2的源代码,描述AlphaFold2是基于神经网络的全新设计的AlphaFold版本,其预测的蛋白质结构能达到原子水平的准确度[56](图5).随后,该团队用AlphaFold完成了98.5%的人类蛋白质的结构预测,并且对其中35.7%的蛋白质结构预测达到了极高准确度[57].AlphaFold2的成功可归因于其神经网络的架构和参考了实验方法解析的蛋白质三维结构的训练程序[58],即基于深度学习的蛋白质结构预测方法,这在结构生物学界引发了巨大的冲击[58].同年7月,DAVID BAKER领导一支计算生物学家团队,基于AlphaFold深度学习的基础,成功开发一款名为RoseTTAFold的工具[59],能够根据有限的信息快速准确地预测出目标蛋白质的结构,达到与AlphaFold2不相上下的准确度.RoseTTAFold所需的计算耗能与计算时间均比AlphaFold2低,仅用一台游戏计算机,在短短十分钟内就可以可靠地计算出蛋白质结构.2021年11月,Science杂志公布了2021年的年度科学突破榜单,AlphaFold和RoseTTAFold两种基于人工智能预测蛋白质结构的技术位列榜首[60],这充分肯定了蛋白质结构预测的光明前景.
虽然人工智能预测蛋白质三维结构有着传统实验方法无法企及的优势,如耗时短、无需对蛋白质进行结晶、无需使用昂贵的实验仪器及材料进行研究;以及源代码的免费开放意味着它是真正适合所有人的蛋白质预测模型,但仍不能否定实验方法的重要性.因为首先需要通过实验方法积累足够多的蛋白质三维结构,才能使得基于算法的AI结构预测有充分的前提条件进行学习模拟;其次,AlphaFold预测蛋白质结构也并非全能的,对于数据库中未能解析的蛋白质柔性部位,使用软件也不能获得准确的结构.AI所做的工作还处于科学实验的辅助阶段,它的可信度还需要实验来证实.
蛋白质的结构预测将有助于为基础生物学提供新的见解,特别对理论假说的提出具有启示和借鉴意义[15],此外还有助于揭示具有临床意义的新药靶点[1],它会与X射线衍射、核磁共振和冷冻电子显微镜等实验方法一起,成为解析蛋白质结构的强大工具,在科学的道路上推进对人类自身的认识和对生命的理解.
5 展 望
人们对于蛋白质结构的深入研究对揭示生命体生化反应的发生机制、研发靶向性药物有着重要意义.X射线衍射晶体学、核磁共振、冷冻电子显微镜三大实验分析技术对于蛋白质结构解析的重要性无需赘言,并且随着科学技术的不断创新,各项技术的壁垒逐渐被打破,彼此之间的界限也变得越来越模糊,这无疑是令人振奋的现象,预示着蛋白质结构解析方法高速发展时期的到来.三大实验技术与AlphaFold人工智能系统相辅相成,已经成为蛋白质结构解析领域最强有力的工具,在未来也必将引领结构生物学的发展,向人们揭示生命的奥秘.
猜你喜欢
机电安全(2022年5期)2022-12-13
中华实验眼科杂志(2022年1期)2022-11-15
中华实验眼科杂志(2021年10期)2021-04-17
科学(2020年1期)2020-01-06
浙江大学学报(理学版)(2016年6期)2016-12-15
分析测试学报(2015年3期)2016-01-13
中国卫生(2015年12期)2015-11-10
物理实验(2015年9期)2015-02-28
中国药业(2014年17期)2014-05-26
电子设计工程(2014年8期)2014-02-27
