
HPLC法检查丙胺卡因原料药有关物质方法研究
2023-07-10 06:52:56陈慧芳
牡丹江医学院学报 2023年3期
王 坤,陈慧芳,杨 莹,马 允
(安庆医药高等专科学校,安徽 安庆 246052)
丙胺卡因(procaine)是一种酰胺类局部麻醉药物,其针剂临床上用于硬膜外、阻滞和润等各种手术麻醉[1-3]。此外常和利多卡因联合制成复方利多卡因乳膏,用于针穿刺,浅层外科手术等皮层局部麻醉[4]。丙胺卡因的作用机制是阻滞神经冲动产生和传导所需的离子流而稳定神经细胞膜,从而产生局部麻醉效应[5]。丙胺卡因原料用于各种局麻制剂的原料药,临床应用广泛,其原料药有关物质各国药典收载均有欠缺,2020年版《中国药典》暂未见收载其有关物质[6-7]。本研究参考欧洲药典10.0,并对药典收载方法进行优化,结合合成工艺得知丙胺卡因原料药共7个已知杂质(A~G,结构见下图1),其中杂质B既为工艺杂质也为降解杂质,其他均为合成工艺杂质。同时丙氨卡因是一种水溶性药物[8],因此在其杂质检查中,使用高效液相色谱法可以实现对水溶性杂质的准确检测和分离,有较高的灵敏度和可靠性。相比之下,气相色谱法主要适用于描绘挥发性化合物,对于水溶性杂质的分析能力较弱,对保障药物质量的检测准确性有一定影响[9]。因此,高效液相色谱法(HPLC)在丙氨卡因杂质检查中具有明显的优势,是首选的检测方法。基于此,本研究建立了专属性强、灵敏度高、准确度高、精密度好有关物质检测方法,适合对丙胺卡因原料药有关物质进行质控。

图1 7种已知杂质
1 仪器与试剂
1.1 仪器设备XS204DR型电子分析天平(瑞士梅特勒公司);ME205E千分之一电子天平(瑞士梅特勒公司);FE20 pH计(瑞士梅特勒公司);Wates e2678高效液相色谱仪(美国Wates公司);HAS超声仪(上海一恒公司)。EMPOWER分析工作站。
1.2 试剂或试药磷酸氢二钠二水合物为分析纯(国药集团公司);磷酸二氢钠一水合物为分析纯(国药集团公司)。丙胺卡因对照品(中检院);杂质A、杂质B、杂质C、杂质D、杂质E、杂质F、杂质G对照品,LGC。丙胺卡因原料(印度)。纯化水;乙腈为HPLC级(阿拉丁公司);磷酸、氢氧化钠均为分析纯(国药集团公司)。
2 方法与结果
2.1 EP10.0方法复核色谱条件,采用封端色谱柱 Waters Xbridge C18(150 mm×4.6 mm,5 μm),以磷酸盐缓冲液(0.18 g磷酸二氢钠和2.89 g磷酸二氢钠二水合物加100 mL水溶解)-乙腈(74∶26)为流动相,柱温25 ℃,流速每分钟1.0 mL,检测波长:240 nm,进样量20 μL。空白溶剂即流动相。系统适用性溶液:取丙胺卡因、杂质A、杂质B、杂质C、杂质D、杂质E、杂质F、杂质G对照品适量,精密称定,用流动相稀释溶解成每毫升约含丙胺卡因2.5 mg、杂质A 2.5 μg、杂质B 0.25 μg、杂质C 2.5 μg、杂质E 2.5 μg、杂质F 2.5 μg与杂质G 2.5 μg的溶液。取空白、系统适用性溶液照色谱条件,注入色谱仪,记录色谱图。见下图2系统适用性溶液图谱。由图谱2数据显示杂质F与杂质A分离度为1.10,
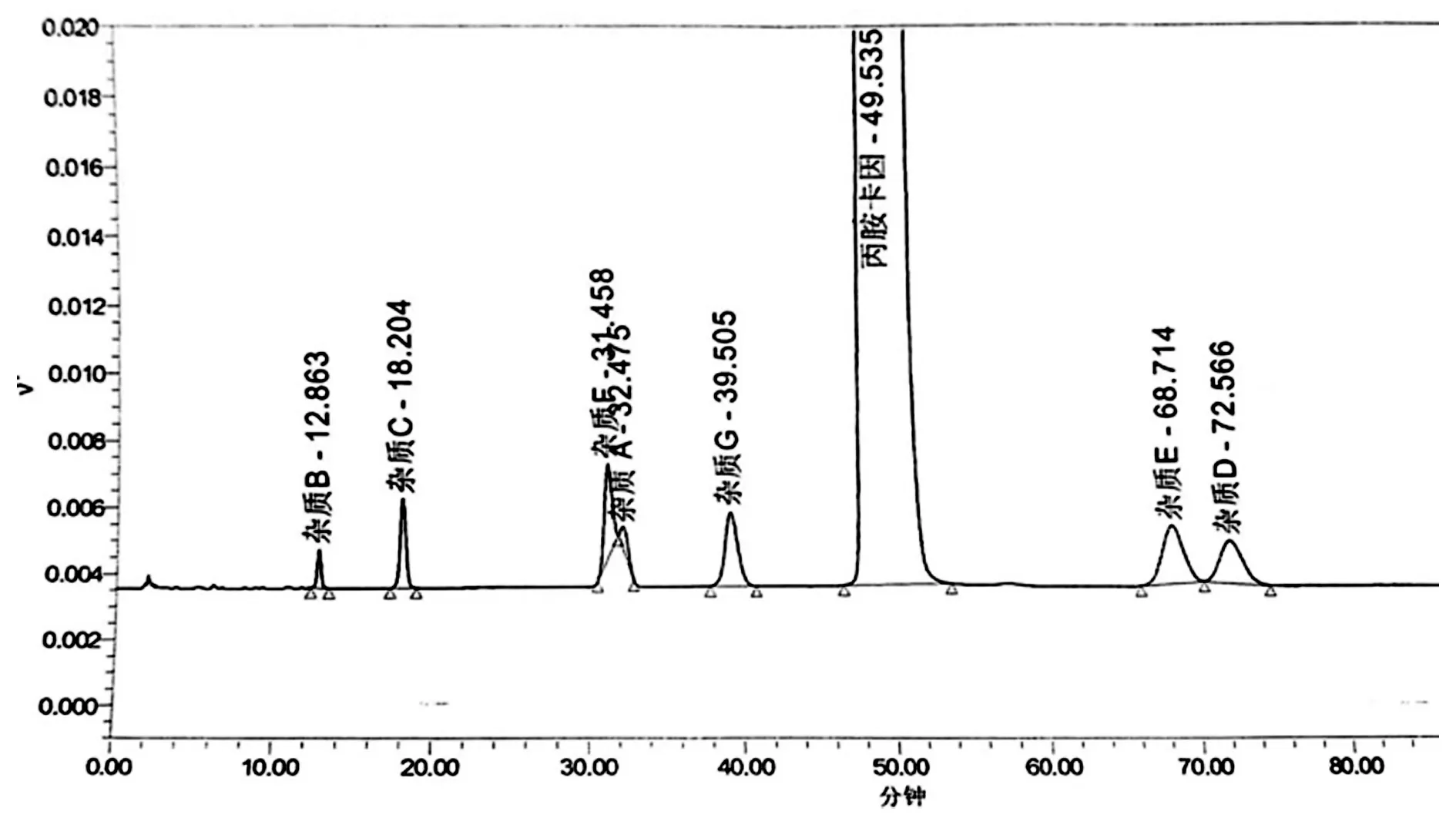
图2 系统适用性色谱图
杂质E与杂质D分离度为1.38,分离度均小于1.5,EP10.0方法不适合丙胺卡因有关物质检测,尝试调整流动相比例等措施对色谱条件进行优化。
2.2 方法优化
2.2.1 流动相比例调整 通过调整相与有机相的比例对EP10.0色谱条件进行优化。(1)增加有机相比例:以磷酸盐缓冲液-乙腈(70∶30)为条件,其他色谱条件不变。取系统适用性溶液注入色谱仪,记录图谱。如下图3所示,系统适用性中杂质A与杂质G可达到完全分离,分离度为2.84,但杂质E与杂质D分离度无明显改善,未达到基线分离,分离度为1.41。(2)降低有机相比例:以磷酸盐缓冲液-乙腈(75∶25)为条件,其他色谱条件不变。此时系统适用性,杂质A与杂质G可达到完全分离,分离度为5.01,但丙胺卡因出峰时间可达到101.116 min,运行时间过长,此色谱条件不合适,如下图4所

图3 增加有机相比例系统适用性

图4 降低有机相比例系统适用性
示。综上所述杂质A与杂质G的分离很好实现,可通过增加有机相比例。但杂质E与杂质D分离通过调整流动相比例对其影响不大。考虑调整水相pH优化色谱条件。
2.2.2 水相pH调整 在增加有机相比例的基础上,分别调整水相的pH至7.0和9.0。水相比例调整至7.0和9.0时杂质E与杂质D的分离度均不改善,此外杂质A与杂质G无法分离。如下图5和下图6所示。通过调整pH发现杂质A与杂质G在降低pH和升高pH情况下,均无法分离,且杂质E与杂质D分离状况未得到改善,综上所述,调整水相pH条件不合适。考虑更换固定相,即更换色谱柱进行实验。
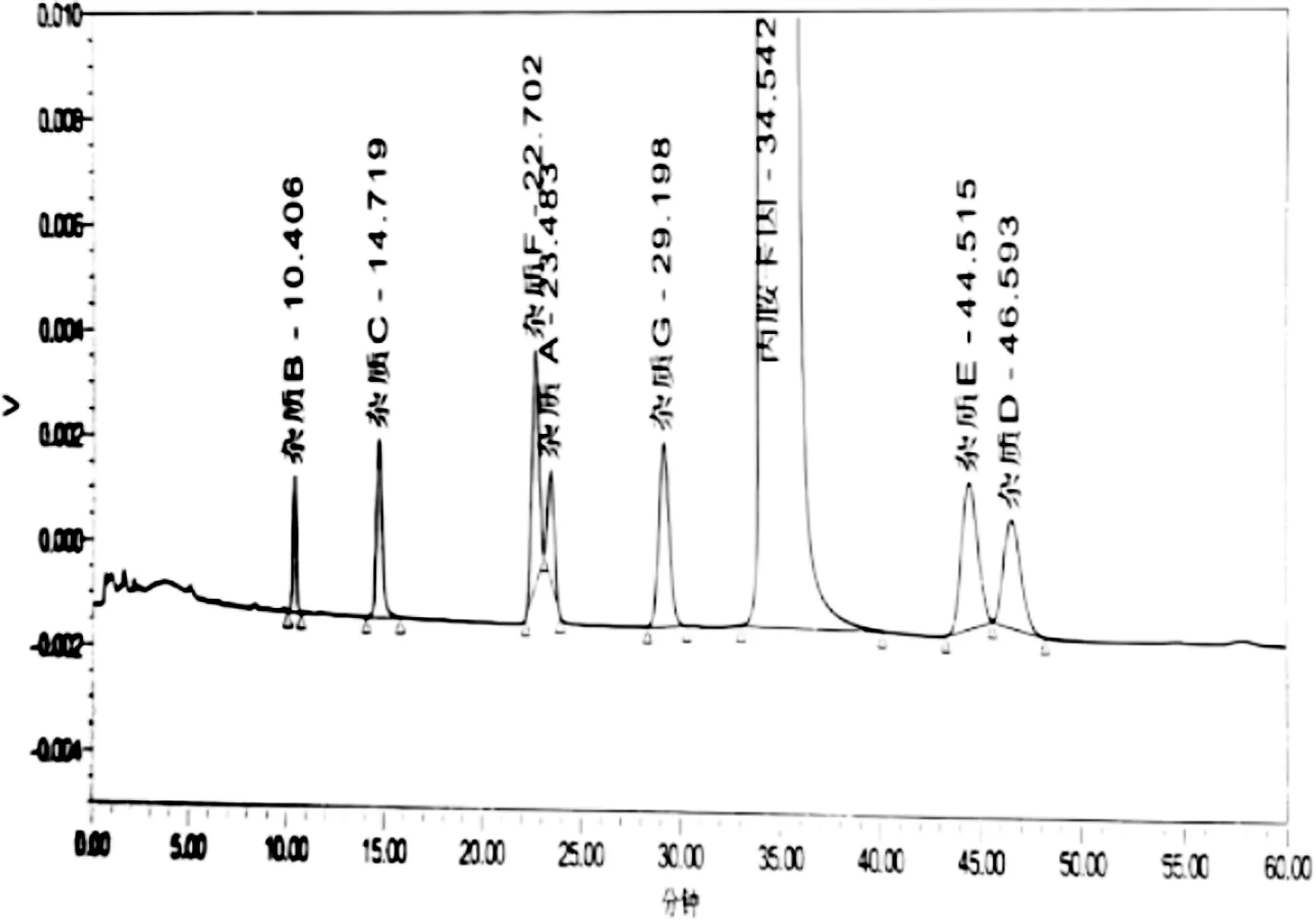
图5 水相7.0系统适用性

图6 降低水相9.0系统适用性
2.2.3 更换色谱柱 通过更换不同品牌色谱柱,最终筛选到两根色谱柱可实现系统适用性中主峰与各杂质之间,及杂质与杂质间的分离。(1)色谱柱1:YMC-Pack ODS-AM C18(150×4.6 mm,5 μm);(2)色谱柱2:Waters Xbridge BEH C18(150×4.6 mm,5 μm)。色谱柱1 杂质E与杂质D分离度为2.19,但主峰出峰时间较晚。色谱柱2杂质E与杂质D分离度为2.17,主峰出峰时间较早。综合考虑色谱柱2较适合丙胺卡因原料有关物质的分析。详见下图7和图8。
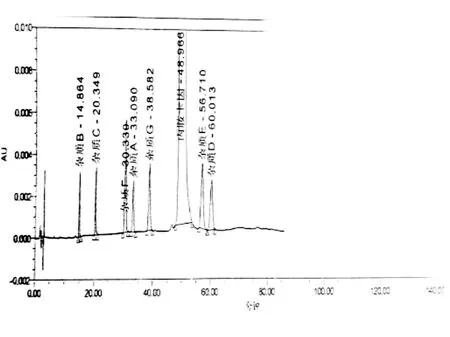
图7 色谱柱1下系统适用性
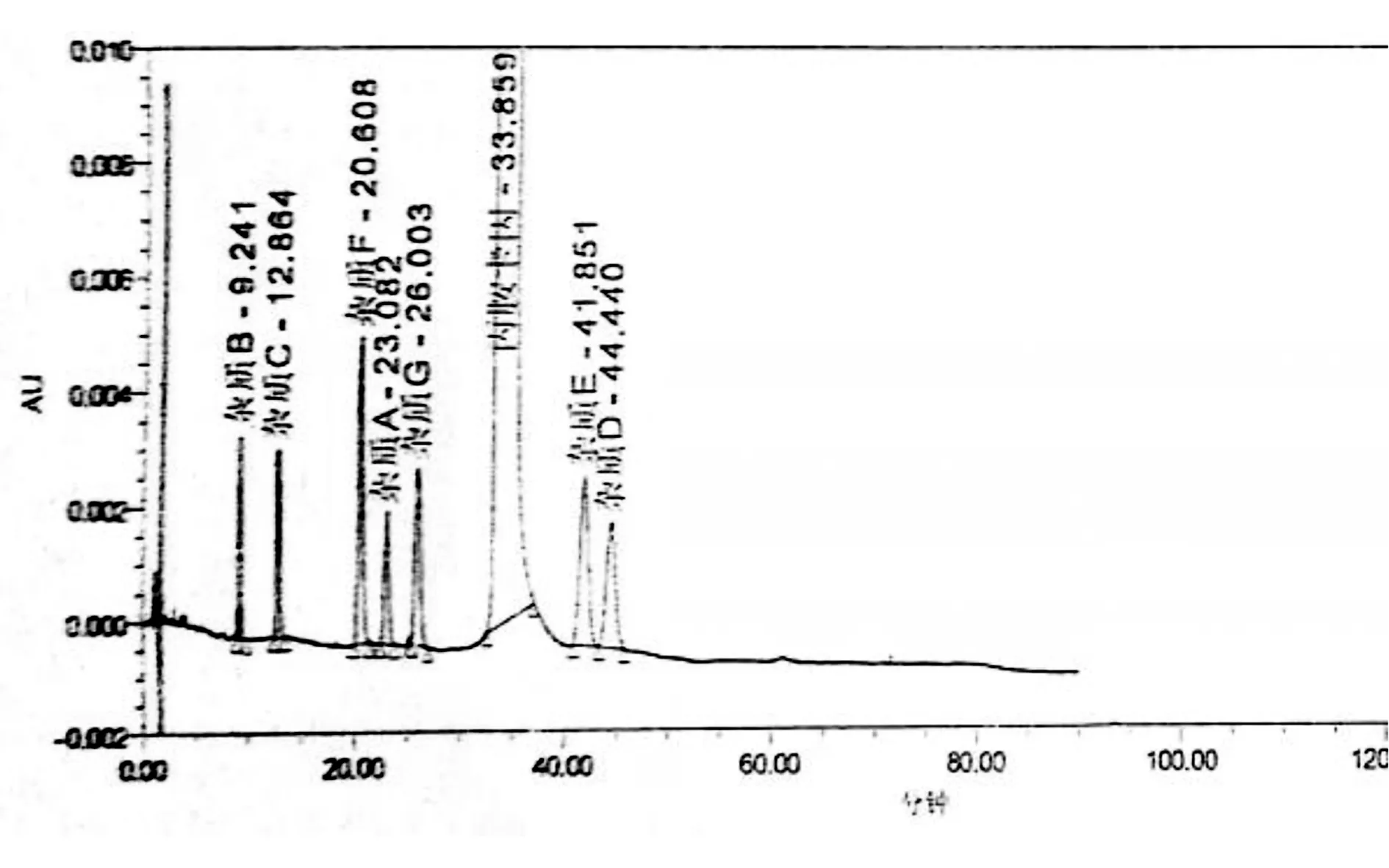
图8 色谱柱2下系统适用性
2.3 分析方法验证根据方法开发结果最终确定丙胺卡因原料药有关物质分析检测方法。具体方法如下。色谱条件:色谱柱,Waters Xbridge BEH C18(150×4.6 mm,5 μm);流动相,磷酸盐缓冲液(0.18 g磷酸二氢钠和2.89 g磷酸二氢钠二水合物加100 mL水溶解)-乙腈(70∶30)为流动相;柱温25 ℃;流速1.0 mL/min;检测波长240 nm;进样体积20 μL。
供试品溶液配制:取丙胺卡因原料适量,精密称定,用流动相稀释溶解成每1 mL约2.5 mg的溶液。系统适用性溶液:取丙胺卡因、杂质A、杂质B、杂质C、杂质D、杂质E、杂质F、杂质G对照品适量,精密称定,用流动相稀释溶解成每毫升约含丙胺卡因2.5 mg、杂质A 2.5 μg、杂质B 0.25 μg、杂质C 2.5 μg、杂质E 2.5 μg、杂质F 2.5 μg与杂质G 2.5 μg的溶液。对照品溶液:取杂质B对照品适量,用流动相稀释成每1 mL约0.25 μg的溶液。自身对照溶液:精密量取供试品溶液适量,用流动相稀释成每1 mL约丙胺卡因2.5 μg的溶液。可接受标准:系统适用性各成分分离度均大于1.5,主峰与杂质E的分离度不得小于3.0。分析方法的专属性、灵敏度、准确度、精密度均应符合《中国药典2020版四部通则》相关规定。
2.3.1 专属性 取空白溶剂(流动相)、系统适用性溶液、供试品溶液照色谱条件注入色谱仪,记录图谱。结果显示溶剂对供试品溶液有关物质测定无干扰。系统适用性符合要求。
2.3.2 检出限、定量限 取杂质A~G对照品、丙胺卡因对照品适量,精密称定,置不同量瓶中,用流动相稀释成每1 mL约上述对照品约1 mg的溶液,作为对照品储备液。取对照品储备液用流动相逐级稀释,并注入色谱仪,取S/N约为10的浓度溶液,作为定量限,取S/N约为3的浓度溶液,作为检出限。详见下表1。结果显示各杂质及主成分分离度均在限度浓度的10%以下,方法灵敏度好。
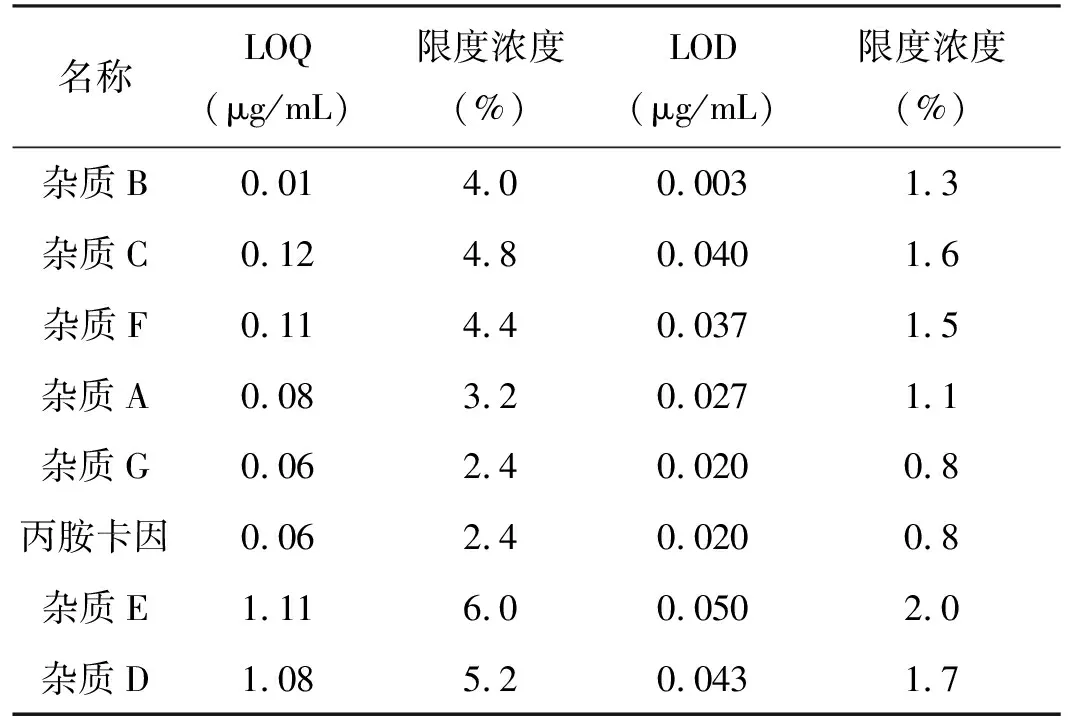
表1 丙胺卡因有关物质定量限、检出限结果
2.3.3 线性与范围 分别取杂质A~G对照品、丙胺卡因对照品适量,精密称定,配制混合对照品储备液,精密量取混合对照品储备液适量,配制约相当于定量限溶液的线性1、50%限度浓度溶液、80%限度浓度溶液、100%限度浓度溶液、120%限度浓度溶液、150%限度浓度溶液与200%限度浓度溶液,以浓度C(μg/mL)为横坐标,以峰面积A为纵坐标,作线性回归方程,查看各杂质与丙胺卡因线性情况。详见下表2。由表2可知杂质A~G与丙胺卡因从定量限~200%限度范围内线性良好,线性相关系数r均大于0.998,杂质B相对校正因子为0.48,其他成分相对校正因子均在0.9~1.1范围内,所有成分截距占比均在25%以内。杂质B为基因毒性杂质且相对校正因子小于0.8,按外标法计算,其他杂质及未知单杂按自身对照法计算。

表2 杂质A~G和丙胺卡因的线性关系及相对校正因子情况
2.3.4 精密度 照验证方法,配置对照品溶液、供试品溶液、对照溶液,其中供试品溶液与自身对照溶液平行配置6份,其中对照品溶液连续进样5针,供试品溶液、自身对照各进1针,杂质B按外标法计算,其他单杂按自身对照计算。6份供试品溶液均只有杂质B、杂质G,其杂质B结果平均分别为0.002 1,杂质G的平均值为0.040,RSD分别为1.96%、1.89%。检出结果的RSD均小于2.0%,方法精密度良好。
2.3.5 准确度 照验证方法,配置对照品溶液,配制对照溶液。取杂质A、C~G对照品约10 mg,精密称定,置10 mL量瓶中,用流动相稀释溶解至刻度,摇匀得混合储备液1。取杂质B约10mg,精密称定,置10 mL量瓶中,用流动相稀释溶解至刻度,摇匀得杂质B储备液。精密量取1.0 mL混合储备液1和0.1 mL杂质B储备液置100 mL量瓶中,用流动相稀释至刻度,摇匀,得混合储备液。80%回收率溶液(n=3):取丙胺卡因原料25 mg,精密称定,置10 mL量瓶中,加2.0 mL混合储备液1,用流动相吸收至刻度,摇匀即得。100%回收率溶液(n=3):取丙胺卡因原料25 mg,精密称定,置10 mL量瓶中,加2.5 mL混合储备液1,用流动相吸收至刻度,摇匀即得。120%回收率溶液(n=3):取丙胺卡因原料25 mg,精密称定,置10 mL量瓶中,加3.0 mL混合储备液1,用流动相吸收至刻度,摇匀即得。取空白、对照品溶液、自身对照、回收率溶液进样测定。测得回收率,均符合《中国药典2020版四部通则》规定。详见下表3。
2.3.6 样品结果测定 取本品以“2.1”项下供试品溶液配制方法配制溶液及采用“2.3”项下的色谱条件检测,再以自身对照加校正因子检测有关物质,检测结果见“2.3.4”,检出结果的RSD均小于2.0%。
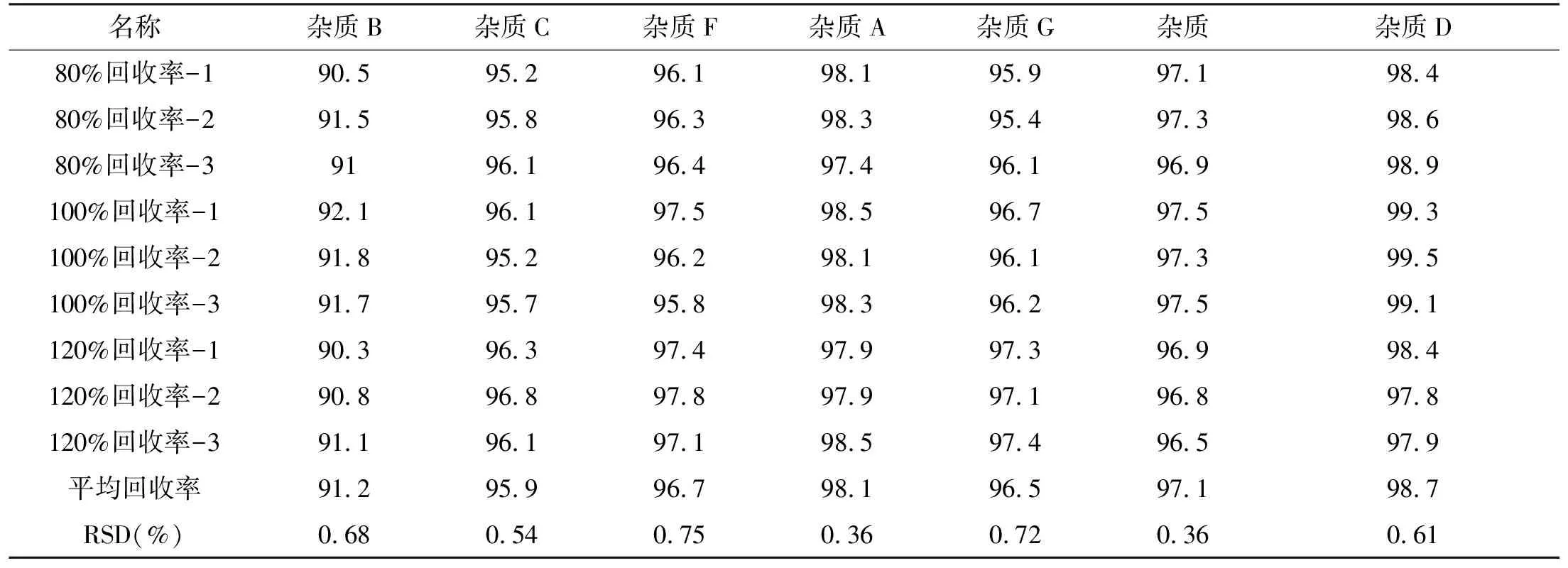
表3 回收率测定结果汇总(%)
3 讨论
3.1 流动相比例及pH的考察在分析方法开发阶段,通过复核EP10.0标准条件发现,杂质F与杂质A,杂质E与杂质D均无法基线分离。在原条件基础上通过调整流动相比例和改变流动相pH发现,调整流动相比例及pH对杂质F与杂质A的分离趋势有明显影响,对杂质E与杂质D无影响,通过比例和pH最终选择水相-有机相(70∶30)对杂质杂质F与杂质A的效果最好,故确定流动相中水相与有机相的比例为70∶30。
3.2 固定相的考察由于调整水相pH条件下杂质A与杂质G在降低pH和升高pH情况下,均无法分离,通过更换不同品牌的色谱柱进行实验,最终通过查阅相关参考文献,最终发现YMC-Pack ODS-AM C18(150×4.6 mm,5 μm)和Waters Xbridge BEH C18(150×4.6 mm,5 μm)对杂质E与杂质D这两种同分异构体化合物分离效果明显。综合分析效率考虑,最终选择Waters Xbridge BEH C18(150×4.6 mm,5 μm)此根色谱柱。
3.3 分析方法验证参考《中国药典2020版四部通则》及《ICH指导原则Q2(R1)》对现有方法进行验证。研究对分析方法关键项进行验证,主要包括专属性、灵敏度、线性范围、重复性、回收率进行验证。经验证后,相关指标均符合方法学验证中规定。与王庆等[10]研究结果类似,表明本方法具有专属性强、灵敏度高、精密度好等优点,可将该方法逐步用于实际工作中。
3.4 研究不足之处研究未对杂质来源未进行更深一步研究,未通过酸、碱、氧化及光照破坏实验,进一步证实杂质是否为降解产生。未进行破坏实验考察分析方法的专属性。未对中间精密度、重现性进行验证。未对供试品溶液及对照品溶液稳定性考察。后续实验将对专属性、精密度、溶液稳定性进一步验证。此外,方法优化中对水相pH调整、流动相比例调整只选择两个水平,后续会继续再增加3个水平进行研究。
综上所述,由于丙胺卡因原料药的检测方法在中国药典中并未收录,本次硏究参考欧洲药典中的方法重新开发并验证了HPLC法检测丙胺卡因有关物质的分析方法,目的在于寻找一种更加科学且准确的检测方法,便于日常的检验或科研工作顺利进行。方法学验证表明,方法的专属性强、灵敏度高、准确度高、精密度好,可满足丙胺卡因原料药有关物质检测需求。相对于药典及文献收集的方法,本次研究的分析方法具有分析时间短,通用性高,可用于丙胺卡因原料的合成工艺中已知单杂的控制。
猜你喜欢
化工设计通讯(2021年8期)2021-08-23 11:49:36
家庭影院技术(2021年2期)2021-03-29 07:18:28
石油地质与工程(2019年3期)2019-09-10 08:27:54
建材发展导向(2019年10期)2019-08-24 06:24:36
淄博师专论丛(2019年1期)2019-04-04 06:01:38
中国钼业(2019年2期)2019-01-19 15:54:06
化工设计通讯(2017年6期)2017-06-26 11:53:37
天津药学(2016年5期)2017-01-16 10:11:26
水利技术监督(2016年6期)2017-01-15 14:01:33
铁道通信信号(2016年1期)2016-06-01 12:10:17
