
UGT1A1基因突变与多例延迟晚发特异性黄疸探索研究*
2023-07-05 10:38:44龚伟邱里吴秀
罕少疾病杂志 2023年6期
龚 伟 邱 里 吴 秀
娄底市妇幼保健院/娄底市儿童医院 (湖南 娄底 417000)
新生儿延迟晚发性黄疸在临床中儿科常见疾病之一,近年来医学认为尿苷二磷酸葡萄糖醛酸转移酶1A1(uridine diphos phate glucuronosyl transferase 1A1,UGT1A1)基因突变与新生儿延迟晚发性黄疸高发生有关。UGA1A1是人体内唯一一种催化胆红素进行结合发生反应的酶,UGA1A1特异性底物为胆红素,UGA1A1在胆红素代谢过程中起到极为重要的作用,UGA1A1基因编码区多态性会引起基因表达或者酶活性变化,与新生儿晚发特异性黄疸高发生率存在密切联系[1]。同时,UGA1A1基因突变会引发Gilbert综合征(先天性非结合型高胆红素血症),此类疾病的发生会增加新生儿期迁延性黄疸的发病风险[2]。本研究旨在探讨UGT1A1基因突变与多例延迟晚发特异性黄疸的关系,现报告如下。
1 资料与方法
1.1 一般资料实验研究选取我院2022年2月1日至2023年3月1日期间临床诊断70例迁延性新生儿黄疸为观察组,65例无黄疸新生儿为对照组,所选新生儿均足月出生,出生后均接受母乳喂养。观察组新生儿胎龄>37周,平均胎龄(275.21±7.62)d;出生时体重>2.5kg,平均体重(3.07±0.62)kg;对照组新生儿为同期住院患儿,胎龄>37周,平均胎龄(276.34±7.21)d;出生时体重>2.5 kg,平均体重(3.11±0.59)kg。两组新生儿在性别、胎龄、体重等项目上差异无统计学意义(t检验,P>0.05)。
观察组纳入标准:年龄在14d~28d;黄疸发生在新生儿时期;黄疸病症持续≥14d且迁延不退;签署研究知情同意书。排除标准:出生后14 天内临床诊断治疗有明确病因导致黄疸迁延不退者;肝细胞性黄疸患儿;阻塞性黄疸患儿;感染性黄疸患儿;母乳性黄疸患儿。
对照组纳入标准:年龄在14d~28d;无病理性黄疸;因新生儿脐炎、肺炎等除黄疸外病因住院;正常新生儿。排除标准:血常规、胸片、C反应蛋白、网织红细胞、肝功能等项目检查有异常;未签署研究知情同意书。
1.2 基因检测方法通过伦理委员会审核及监护人知情同意情况下,对符合条件的新生儿进行UGT1A1基因Gly71Arg位点进行检测,方法如下:(1)基因组DNA提取:使用EDTA抗凝管采集新生儿外周血2mL,使用试剂盒(fastPure blood DNA Isolation mini kit V2,vazyme)提取DNA,基因组DNA 4℃保存。(2)靶向引物设计:以UGT1A1基因的侧翼序列和第一外显子设计引物。(3)PCR:反应体积为20μL,采用TaKaRa rTaq聚合酶进行扩增,预变性95 ℃、5 min,变性 94 ℃、30 s,退火60 ℃、40 s,延伸72 ℃、1min,循环35次,最后延伸72 ℃、10min。将PCR产物置于1.5%琼脂糖凝胶进行电泳并溴乙锭染色,通过透射式紫外线灯对产物片段大小进行观察。
1.3 统计方法本研究所有数据均纳入SPSS 22.0进行统计分析,计数资料以[n,(%)] 表示行χ2检验,计量资料行t检验,以(±s) 表示平均数±标准差,以α=0.05为标准,若P<0.05为有统计学意义。
2 结 果
2.1 两组新生儿一般资料比较见表1。
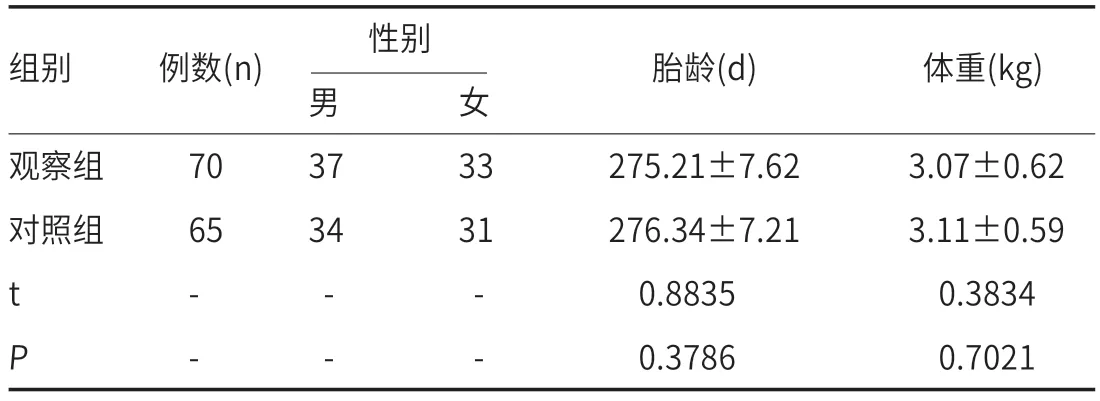
表1 两组新生儿一般资料
2.2 两组UGT1A1基因突变比较见表2。两组新生儿基因型分布存在差异(卡方检验,P<0.05),突变型(纯合子和杂合子)观察组34(48.57%)明显高于对照组16(24.62%),P<0.05;Arg等位基因频率观察组29.29%明显高于对照组13.85%(卡方检验,P<0.05);Gilbert综合征发病风险观察组42(60.00%)明显高于对照组12(18.46%),P<0.05。

表2 两组UGT1A1基因突变 [n,(%)]
2.3 两组新生儿血清总胆红素均值比较见表3。两组新生儿血清总胆红素比较中,由于新生儿出生72h内,在不同时间的总胆红素干预黄疸的数值并非固定不变,因此选择出生72h后,干预黄疸的总胆红素值作为峰值进行比较为准,观察组70例新生儿72 h后总胆红素均值396.37±79.15μmol/L明显高于对照组65例新生儿总胆红素均值305.21±41.34μmol/L(t检验,P<0.05)。
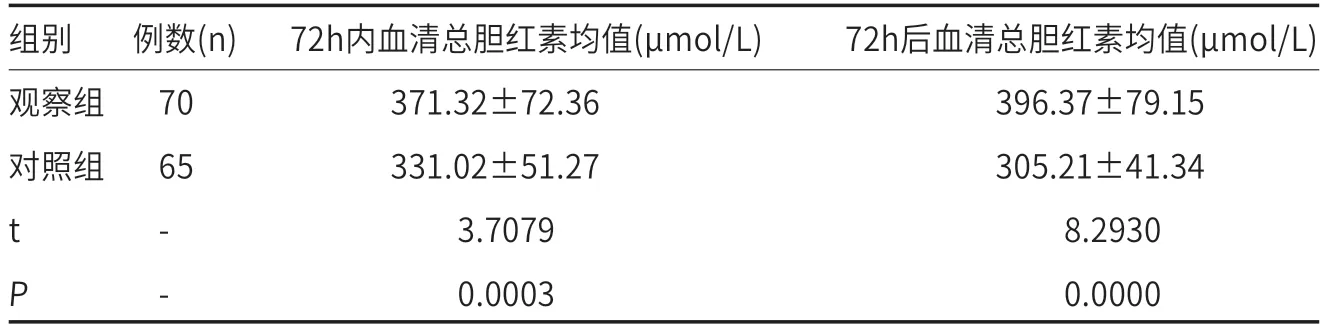
表3 两组新生儿血清总胆红素均值
3 讨 论
新生儿黄疸是新生儿时期常见的高发病症,也是全球范围内儿科医生共同关注的热点和重点问题[3]。目前临床常规的诊疗方法对新生儿黄疸已经不能获取到较为满意的治疗效果,甚至部分黄疸重症新生儿死于急性期,幸存患儿有七成以上可能并发较为严重的神经系统后遗症,对新生儿生存质量及健康情况造成极为严重的威胁[4]。近年来有资料显示尿苷二磷酸葡萄糖醛酸转移酶(UGT)基因的变异与多态性是有可能导致新生儿黄疸的遗传高危风险因素。UGT基因存在于部分脊椎动物的肝部微粒体中,是糖基转移酶超家族之一,同时也是生物体内进行Ⅱ相生物转化过程中最重要的酶之一[5]。其中UGT1A1基因是肝脏中刺激胆红素葡萄糖醛酸化产生反应的活性酶,也是对胆红素清除起到调节作用的关键酶[6]。也有研究表明新生儿体内会产生较多的胆红素,但由于其胆红素清除效率较低,部分未发生结合反应的胆红素进入肝细胞当中,在UGT1A1的影响下与葡萄糖醛酸产生反应,导致水溶性胆红素生成,后随尿液排出,降低新生儿体内胆红素含量。当UGT1A1基因发生突变,其结构出现变化,原本功能随之缺失,造成胆红素结合能力发生下降,从而引发新生儿黄疸病症[7-8]。Gilbert综合征属于一种较为常见的遗传性肺结核高胆红素血症,此病症的发生与UGT的活性下降有着密切的关系。临床研究表明控制Gilbert综合征发病的UGT基因在2号染色体2q37上,正常情况下UGT基因编码的蛋白会使葡萄糖醛酸与胆红素相结合,使得非结合胆红素转为结合性胆红素[9-11]。当UGT1A1基因发生变异后,基因的表达水平下降导致非结合胆红素转化的过程受到影响,最终导致Gilbert综合征的发生,同时Gilbert综合征也是促使新生儿发生迁延性黄疸病症发病风险的重要因素[12-13]。
UGT1A1基因编码序列变异会导致UGT结构发生异常,出现酶结合功能方面的丧失与缺陷,而在启动子区域的核苷酸多态性会导致基因表达能力明显下降,致使酶的活性降低,使得非结合胆红素在人体体内蓄积,最终成为引发新生儿黄疸的高危因素。在临床实验中已明确UGT1A1的基因突变有两种类型,TATA插入型和编码区突变型,后者又分为Gly71Arg、Arg367Gliy等不同基因型,本次研究表明新生儿黄疸与UGT1A1基因Gly71Arg的突变存在密切关系,且该突变对延迟晚发特异性黄疸的发生有一定的影响作用。本次实验研究结果表明,在参与实验的两组新生儿中,在迁延性黄疸患儿突变型(纯合和杂合)34例(48.57%)相明显高于对照组无黄疸病症新生儿16例(24.62%),同时等位基因(Arg)的频率观察组(29.29%)也明显要高于对照组(13.85%),这表明UGT1A1基因的Gly71Arg点突变与迁延性黄疸的发生有着较强的关联性。参与实验的两组患儿中,Gilbert综合征潜在发病风险观察组42(60.00%)较对照组12(18.46%)明显更高,这也表明Gilbert综合征会影响新生儿迁延性黄疸的发病风险,带有Arg等位基因突变的新生儿出现迁延性黄疸的风险会呈正比增加。本次研究对象当中,发生UGT1A1基因Gly71Arg点突变的患儿血清总血胆红素水平明显要高于无迁延性黄疸患儿,说明Gly71Arg突变会导致新生儿体内血胆红素提高,这种升高可能是葡萄糖醛栓中转移酶活性被影响而造成的。在本次研究中未包括不足月新生儿,主要因其特殊的生理特点,若患有黄疸一类病症则表现较为严重,耐受性差,而且极易引发机体神经系统损伤。
通过本次研究结果可以看出,UGT1A1基因的突变与新生儿延迟晚发性黄疸有着极为紧密的联系,其引发的Gilbert综合征也会导致新生儿延迟晚发性黄疸发病风险上升,UGT1A1基因多态性研究具有十分重要的临床意义,其所发生的基因突变现象与多种疾病相关,也会影响多种药物代谢过程。虽然新生儿黄疸是新生儿期较为常见的病理现象,但若发展成高胆红素血症,会严重威胁患儿的生命安全并造成严重或永久性的精神系统损伤而产生后遗症,所以及时准确的寻找病因,对此类疾病进行积极预防,早期治疗和干预显得极为重要。
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
肝博士(2022年3期)2022-06-30 02:48:34
昆明医科大学学报(2021年3期)2021-07-22 07:39:56
基层中医药(2021年10期)2021-06-05 07:15:28
中华养生保健(2020年10期)2021-01-18 06:46:06
中国生殖健康(2020年2期)2021-01-18 02:51:26
肝博士(2020年5期)2021-01-18 02:50:26
小学生导刊(2018年13期)2018-06-29 03:49:00
临床医药文献杂志(电子版)(2017年11期)2017-05-17 04:48:28
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
