
CTAB 添加量对硅-磷灰石成分和形貌的调控研究
2023-06-30 05:23金莹邓繁艳宁聪琴
上海师范大学学报·自然科学版 2023年1期
关键词:表面活性剂
金莹 邓繁艳 宁聪琴


摘要:磷酸钙是人体骨组织无机质的主要成分,而硅(Si)元素被证明具有诱导成骨的作用.因此,含 Si 的磷酸钙作为骨缺损修复材料受到了广泛的研究.以四水合硝酸钙(Ca( NO3)2·4H2 O)、磷酸氢二铵((NH4)2HPO4)和正硅酸乙酯(TEOS )分别为钙(Ca )源、磷(P )源和 Si 源,以十六烷基三甲基溴化铵(CTAB )为表面活性剂,采用水热法合成纳米硅-磷灰石粉体.研究了水热反应中 CTAB 添加量对合成的纳米硅-磷灰石粉体的化学组成、晶体结构和形貌的影响.结果表明:CTAB 的加入不仅能降低纳米硅-磷灰石的颗粒尺寸,还能促进 Si 进入磷灰石晶格,提高硅-磷灰石中 Si 的含量.当 CTAB 添加量为0.55 g 时,合成的硅-磷灰石的物质的量之比 n ( Ca )∶n ( P )∶n ( Si )可达5∶2∶1,此纳米硅-磷灰石粉体经高温煅烧后,更易于形成正交结构的硅磷酸钙(Ca5( PO4)2SiO4,CPS )相.
关键词:硅磷酸钙(CPS );十六烷基三甲基溴化铵(CTAB );表面活性剂;水热合成
中图分类号:O 611.6 文献标志码:A 文章编号:1000-5137(2023)01-0008-07
Influence of CTAB addition on the composition and morphology of Si-apatite
JIN Ying,DENG Fanyan,NING Congqin*
(College of Chemistry and Materials Science,Shanghai Normal University,Shanghai 200234,China)
Abstract:Calcium phosphate is the main component of inorganic matter in human bone tissue,and silicon( Si ) has been proved to have an effect on inducing bone formation. Therefore,silicon-containing calcium phosphate has been widely studied as a bone defect repair material. In this paper,nano Si-apatite powder was synthesized by a hydrothermal method,using calcium nitrate tetrahydrate( Ca ( NO3)2·4H2O ), ammonium phosphate dibasic(( NH4)2HPO4), and tetraethyl orthosilicate(TEOS ) as calcium ( Ca ) source,phosphorus(P ) source,and silicon source,respectively. In addition,cetyltrimethylammonium bromide( CTAB )was used as a surfactant and the effect of CTAB amount on the chemical composition,crystal structure,and morphology of the synthesized Si-apatite powder was studied. The results indicated that the addition of CTAB could not only reduce the particle size of nano Si-apatite ,but also promote Si element entering the apatite lattice ,improving the Si content in Si-apatite. When the addition amount of CTAB is 0.55 g,the mole ratio of n ( Ca )∶n ( P )∶n ( Si ) in the synthesized Si-apatite could reach 5∶2∶1. After calcination at high temperature,this nano Si-apatite powder was easier to form the orthogonal silicon-calcium phosphate ( Ca5( PO4)2SiO4,CPS ) phase .
Key words:silicon-calcium phosphate( CPS );cetyltrimethylammonium bromide( CTAB );surfactant;hydrothermal synthesis
0 引言
磷酸鈣类生物陶瓷,如羟基磷灰石(Ca10( PO4)6( OH )2,HA )和β-磷酸三钙(β-Ca3( PO4)2,β-TCP ),因具有与天然骨组织相似的化学组成而被广泛应用于人体硬组织修复,其不仅具有良好的生物相容性,还可在体内与周围骨组织通过化学反应形成稳定的骨性结合[1-2].然而,HA 和β-TCP 等磷酸钙类陶瓷缺乏骨诱导性,导致新生骨组织生长缓慢.作为人体中重要的微量元素之一,硅(Si )被发现在骨组织生长方面发挥着重要作用[3].CARISLE[4]利用电子微探针技术发现 Si 位于大鼠的活性钙化部位,并通过体内和体外研究表明 Si 对新骨组织形成和矿化有重要影响.为了改善磷酸钙生物陶瓷的生物活性,含 Si 的磷酸钙类陶瓷在近些年来受到了广泛关注,如 Si 掺杂的羟基磷灰石(Si-HA )、Si 掺杂的磷酸三钙(Si-TCP )等.这些含 Si 的磷酸钙材料均被发现具有良好的促成骨能力[5-7].
以往的研究发现,含硅磷酸钙类陶瓷在烧结过程中往往会生成一种新的物相——正交结构的硅磷酸钙(Ca5( PO4)2SiO4,CPS )[7-9].CPS 的钙、磷和硅的物质的量之比 n ( Ca )∶n ( P )∶n ( Si )=5∶2∶1,单位晶胞中钙(Ca )元素和硅+磷(Si+P )的物质的量之比为1∶1.67,与 HA 单位晶胞中 Ca 和 P 元素的物质的量之比相同[10].研究发现,CPS 陶瓷具有良好的生物活性和生物相容性,能在体外显著促进骨髓间充质干细胞的增殖和成骨分化,体内结果显示,CPS 相较于 HA 具有更好的骨修复能力[11-12].前期研究发现,前驱体的成分、结构及尺寸对 CPS 粉体的煅烧温度、成分和物相有显著影响.
粉体的合成方法主要包括固相反应法[13]、溶胶-凝胶法[14]、化学沉淀法[15]和水热合成法[16].其中,水热合成法被广泛应用于纳米粉体的制备,相较于固相反应法,该方法制备的纳米粉体具有分散性佳、晶粒尺寸小、结晶性好等优点[17-18].在水熱反应法中,表面活性剂对纳米粉体的形貌和尺寸具有重要影响.许多研究者利用表面活性剂作为模板剂调控微纳米 HA 的结晶行为,实现对 HA 的晶体形貌和尺寸的控制,并将其应用到药物负载领域[19-20].因此,本文作者利用十六烷基三甲基溴化铵(CTAB )作为表面活性剂,采用水热合成法制备了纳米硅-磷灰石粉体,研究了 CTAB 添加量对硅-磷灰石粉体粒径和形貌的调控规律.
1 材料制备及表征
1.1 材料的制备
1.1.1 实验原料
本实验中所采用的试剂主要包括:CTAB,阿拉丁试剂(上海)有限公司,分析纯(AR );四水合硝酸钙( Ca ( NO3)2·4H2 O ),上海凌峰化学试剂集团公司,AR;磷酸氢二铵((NH4)2HPO4),广州化学试剂公司, AR;正硅酸乙酯(TEOS ),上海凌峰化学试剂集团公司,AR.
1.1.2 纳米硅-磷灰石粉体的合成
分别称取质量为0.11,0.33和0.55 g 的 CTAB 溶解于30 mL 去离子水中(对照组不加 CTAB),将0.29 g Ca( NO3)2·4H2 O 加入上述溶液中,充分搅拌,并用氨水维持溶液的 pH 值在10~11之间.随后加入53.69μL TEOS,并将该混合溶液在室温下搅拌1 h,标记为溶液 A;将0.06 g(NH4)2HPO4溶解于30 mL 去离子水中充分搅拌30 min,记为溶液 B;将溶液 B 逐滴加入到溶液 A 中,充分搅拌1 h,之后将混合溶液转入100 mL 聚四氟乙烯内衬的高压反应釜中,放置于180℃的均相反应器中,水热反应6 h.待高压反应釜冷却至室温,取下并打开高压反应釜,将沉淀物用去离子水和无水乙醇充分洗涤、离心3次.将所得粉体放置于60℃恒温鼓风干燥箱进行干燥处理,得到呈白色粉末状前驱体.最后,将干燥后的粉体置于马弗炉中,以5℃·min-1的速率升至1200℃,煅烧6 h,得到 CPS 粉体.
1.2 材料测试与表征
1.2.1 化学组成成分分析
使用 X 射线荧光定量分析仪(XRF )对前驱体粉体的化学组成进行分析.
1.2.2 物相表征
利用 X 射线衍射仪(XRD )表征水热法合成的前驱体及高温煅烧粉体的物相.测试选用铜(Cu ) Kα射线(波长为1.5418×10-10 m),扫描角度为10°~60°,步长为0.02°,扫描速度为5(°)·min-1.
1.2.3 红外光谱分析
采用傅里叶红外光谱仪分析仪(FRIT )对前驱体粉体中的化学基团进行分析,扫描范围为400~4000 cm-1.
1.2.4 显微形貌表征
使用场发射扫描电子显微镜(FE-SEM )对前驱体及高温煅烧粉体的显微形貌进行观察分析.
2 实验结果与讨论
2.1 粉体物相表征及化学组成成分分析
采用 Ca( NO3)2·4H2O,( NH4)2HPO4和 TEOS 分别作为 Ca 源、P 源和 Si 源,CTAB 作为模板剂.水热反应后,除去杂质并经干燥处理后得到4种不同 CTAB 添加量条件下所制备的硅-磷灰石粉末.为确定并分析硅-磷灰石中 Ca,P 和 Si 元素的含量,利用 XRF 对4种硅-磷灰石粉体的化学成分进行测定,并将 Ca,P 和 Si 的氧化物质量分数转换为对应单质的物质的量之比,结果如表1所示.不同 CTAB 添加量条件下所得前驱体粉末中均有 Si 成分,说明前驱体均为含 Si 的磷灰石.随着 CTAB 添加量的增多,前驱体中 P 元素含量逐渐减小,Si 元素含量明显增多.当 CTAB 添加量增加到0.55 g,P 元素对应的氧化物五氧化二磷( P2O5)质量分数从34.69%减小到30.31%,Si 元素对应的氧化物二氧化硅(SiO2)质量分数从7.21%增加到12.17%,n ( Ca )∶n ( P )∶n ( Si )也逐渐接近 CPS 的5∶2∶1.
为确定不同添加量 CTAB 条件下所得硅-磷灰石粉末的物相组成,对4种硅-磷灰石粉体进行 XRD 表征,结果如图1所示.4种不同 CTAB 添加量条件下所得前驱体粉末的衍射峰均与标准 PDF 卡片 NO.09-0432相吻合,存在(002),(211),(112),(300),(202),(310),(222),(213)和(004)的晶面衍射峰.且 XRD 图谱中无明显杂峰,表明4种不同 CTAB 添加量所得的硅-磷灰石粉末均具有磷灰石结构.
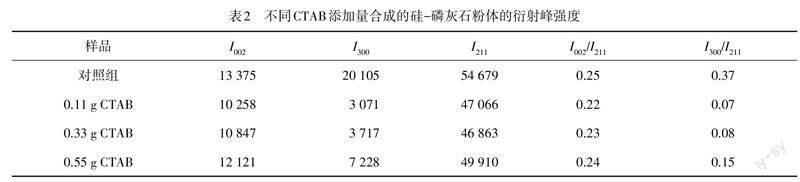
为进一步研究 CTAB 对磷灰石晶体生长的影响,分别计算硅-磷灰石粉末 I002,I211和 I300衍射峰强度,结果如表2所示.CTAB 加入后,硅-磷灰石粉体 I002/I211和 I300/I211峰强比均呈先减小后增大的趋势,且实验组 I002/I211和 I300/I211的峰强比均比未加 CTAB 所得粉末的峰强比更小,表明 CTAB 的加入抑制了硅-磷灰石粉体(002)和(300)晶面的生长,一定程度上阻碍了磷灰石的择优取向生长.这主要是由于 CTAB 带正电的一侧与磷酸根(PO43?)的相互作用会阻碍 HA 生长基元 Ca-P6O24的形成,减缓基元 Ca-P6O24进入 HA 晶格,使磷灰石晶体在 c 轴方向的生长速度变慢,这改变了磷灰石 a,b 和 c 轴上的相对生长速度,进而改变了不同衍射峰的强度比[21-22].此外,硅酸根(SiO44-)可能占据 PO43-位置,并引起 a,b 轴方向的 OH-缺失,导致实验组(300)晶面生长明显被抑制[23].
XRD 和 XRF 結果表明,水热反应可能使 Si 元素成功进入磷灰石晶格,而不是以 SiO2或硅酸钙( CaSiO3)的形式与磷灰石混合.此外,添加 CTAB 后,产物中 Si 含量随着 CTAB 含量的增加而增加,图1中的 XRD 结果显示无其他杂峰,表明 CTAB 能有效促进 Si 元素进入磷灰石的晶格,且进入磷灰石晶格中的 Si 逐渐增多.
将不同 CTAB 添加量条件下所得的硅-磷灰石粉体在1200℃下进行高温煅烧,所得产物的 XRD 谱图如图2所示.当未添加 CTAB 时,1200℃煅烧后粉体的物相以 HA( PDF#09-0432)为主,经 PDF 卡片比对,杂峰的物相可能为羟基硅酸钙(Ca2SiO3( OH )2)( PDF#82-1211).当 CTAB 添加量为0.11 g 时,XRD 结果显示正交结构的 CPS( PDF#40-0393)的特征峰开始出现,随着 CTAB 添加量增至0.33~0.55 g,粉体的主晶相转变为 CPS 相.这说明 CTAB 含量的增加有利于煅烧后的粉体从 HA 相向 CPS 相转变.此外,高温煅烧后的 XRD 结果进一步验证了表1中的 XRF 结果,随着硅-磷灰石中 n ( Ca )∶n ( P )∶n ( Si )逐渐接近5∶2∶1,粉体经煅烧更容易形成 CPS 相.
2.2 粉体 FTIR 分析
选用 FTIR 进一步研究 CTAB 添加量对硅-磷灰石粉体合成的影响,和前驱体在高温煅烧过程中官能团的变化,结果如图3(a)所示.当不添加 CTAB 时,前驱体红外光谱为典型的 HA 峰型,位于630 cm-1和3570 cm-1的吸收峰为羟基的特征吸收峰,位于1100 cm-1和960 cm-1的特征峰为 P— O 伸缩振动峰,位于608,560和470 cm-1的吸收峰对应于 O—P— O 的弯曲振动峰.随着 CTAB 添加量的增大,进入磷灰石晶格中的Si 含量增多,630 cm-1和3570 cm-1位置 OH-的峰强,以及位于1100,960,608,560和470 cm-1,属于 PO43-的吸收峰强度逐渐减弱,表明 SiO44-取代 PO43?进入磷灰石晶格中,为维持电荷平衡从而引起 OH-缺失.760 cm-1处对应于 SiO44-的吸收峰随 CTAB 添加量的增加而逐渐增强.经1200℃高温处理后,CPS 粉体的 FTIR 光谱如图3(b)所示.同样可以观察到,随着 CTAB 添加量的增加,对应于 SiO44-的吸收峰逐渐增强,而对应于 PO43?的吸收峰强度逐渐减弱.由以上结果可推测出:由于 CTAB 的加入,在前驱体的生长过程中,TEOS 水解产生的 SiO44-取代 PO43?进入磷酸钙晶格的比例增加.
2.3 显微形貌分析
为探究在水热反应过程中 CTAB 影响硅-磷灰石粉体组分的机理,通过 FE-SEM 对前驱体粉末微观形貌进行观察.由图4可知,水热反应合成的硅-磷灰石均为纳米颗粒,随着 CTAB 添加量的增多,纳米硅-磷灰石的长度和长径比逐渐变小,粉末由长棒状变为类球状再变为短棒状.如图5所示,在水热条件下,CTAB 作为阳离子表面活性剂可以和 SiO44-以及 PO43-发生静电作用,吸附在负离子配位体生长基元 Ca- Si6 O24和Ca-P6 O24上,或者已形成的磷灰石晶体表面,阻碍磷灰石晶体的生长.同时,CTAB 作为表面活性剂,可有效分散反应物,使溶液中产生更多的形核位点,在降低产物尺寸的同时更有利于Si 进入磷灰石晶格.因此,随着CTAB 添加量的增多,纳米硅-磷灰石粉末中 n( Ca)∶n( P)∶n( Si)也逐渐趋于CPS 中的5∶2∶1.
3 结论
采用 CTAB 为表面活性剂,经过水热反应成功制备 n ( Ca )∶n ( P )∶n ( Si )=5∶2∶1的纳米硅-磷灰石. CTAB 的加入能明显促进 Si 元素进入磷灰石晶格,CTAB 与 SiO44-,PO34-的相互作用可能导致磷灰石晶体生长速度变慢,使纳米硅-磷灰石的形貌由长棒状变为类球状再变为短棒状.此外,CTAB 作为表面活性剂,可有效地分散反应物,使得溶液中产生更多的形核位点,有利于降低纳米硅-磷灰石的尺寸,实现更加均匀的化学反应,从而在煅烧时更容易形成 CPS 相.在本实验中,0.55 g CTAB 添加量条件下所合成的纳米硅-磷灰石颗粒尺寸最小,且 n ( Ca )∶n ( P )∶n ( Si )趋向于 CPS 中的5∶2∶1,是一种潜在的骨修复材料.
参考文献:
[1] HABIBOVIC P,BARRALET J E. Bioinorganics and biomaterials:bone repair [J]. Acta Biomaterialia,2011,7(8):3013-3026.
[2] ELIAZ N ,METOKI N. Calcium phosphate bioceramics:a review of their history ,structure ,properties ,coatingtechnologies and biomedical applications [J]. Materials,2017,10(4):334.
[3] CARLISLE E M. Silicon:an essential element for the chick [J]. Nutrition Reviews,1982,40(7):210-213.
[4] CARLISLE E M. Silicon:a possible factor in bone calcification [J]. Science,1970,167(3916):279-280.
[5] ZHAI W,LU H,WU C,et al. Stimulatory effects of the ionic products from Ca-Mg-Si bioceramics on both osteogenesisand angiogenesis in vitro [J]. Acta Biomaterialia,2013,9(8):8004-8014.
[6] LUGO G J,MAZ?N P,DE AZA P N. Phase transitions in single phase Si-Ca-P-based ceramic under thermal treatment [J].Journal of the European Ceramic Society,2015,35(13):3693-3700.
[7] DELGADO-RUIZ R A,CALVO GUIRADO J L,ROMANOS G E. Bone grafting materials in critical defects in rabbit calvariae:a systematic review and quality evaluation using arrive guidelines [J]. Clinical Oral Implants Research,2018,29(6):620-634.
[8] BIANCO A ,CACCIOTTI I ,LOMBARDI M ,et al. Si-substituted hydroxyapatite nanopowders:synthesis ,thermalstability and sinterability [J]. Materials Research Bulletin,2009,44(2):345-354.
[9] WEI X,AKINC M. Si,Zn-modified tricalcium phosphates:a phase composition and crystal structure study [J]. KeyEngineering Materials,2005,284:83-88.
[10] BALAS F ,PEREZ-PARIENTE J ,VALLET-REGI M. In vitro bioactivity of silicon-substituted hydroxyapatites [J].Journal of Biomedical Materials Research Part A,2003,66(2):364-375.
[11] LU W,DUAN W,GUO Y,et al. Mechanical properties and in vitro bioactivity of Ca5(PO4)2SiO4 bioceramic [J]. JournalofBiomaters Applications,2012,26(6):637-650.
[12] DUAN W,NING C,TANG T. Cytocompatibility and osteogenic activity of a novel calcium phosphate silicate bioceramic:silicocarnotite [J]. Journal of Biomedical Materials Research Part A,2013,101(7):1955-1961.
[13] ARCOS D,RODR GUEZ-CARVAJAL J,VALLET-REG M. The effect of the silicon incorporation on the hydroxylapatitestructure:a neutron diffraction study [J]. Solid State Sciences,2004,6(9):987-994.
[14] BALAMURUGAN A,REBELO A H S,LEMOS A F,et al. Suitability evaluation of sol-gel derived si-substituted hydroxyapatitefor dental and maxillofacial applications through in vitro osteoblasts response [J]. Dental Materials,2008,24(10):1374-1380.
[15] GIBSON I R,BEST S M,BONFIELD W. Effect of silicon substitution on the sintering and microstructure of hydroxyapatite [J].Journal of the American Ceramic Society,2002,85(11):2771-2777.
[16] KIM S R,LEE J H,KIM Y T,et al. Bioactive behaviors of porous Si-substituted hydroxyapatite derived from coral [J]. KeyEngineering Materials,2003,254:969-972.
[17] QI Y,SHEN J,JIANG Q,et al. The morphology control of hydroxyapatite microsphere at high pH values by hydrothermalmethod [J]. Advanced Powder Technology,2015,26(4):1041-1046.
[18] WANG Y ,REN X ,MA X ,et al. Alginate-intervened hydrothermal synthesis of hydroxyapatite nanocrystals withnanopores [J]. Crystal Growth and Design,2015,15(4):1949-1956.
[19] WANG Y,ZHANG S,WEI K,et al. Hydrothermal synthesis of hydroxyapatite nanopowders using cationic surfactant asa template [J]. Materials Letters,2006,60(12):1484-1487.
[20] WU J,ZHU Y J,CHEN F,et al. Amorphous calcium silicate hydrate/block copolymer hybrid nanoparticles:synthesisand application as drug carriers [J]. Dalton Transactions,2013,42(19):7032-7040.
[21] RANJIT K T,KLABUNDE K J. Amphiphilic templating of magnesium hydroxide [J]. Langmuir,2005,21(26):12386-12394.
[22] WIERZBICKI A,CHEUNG H S. Molecular modeling of inhibition of hydroxyapatite by phosphocitrate [J]. Journal ofMolecular Structure:Theochem,2000,529(1/2/3):73-82.
[23] LEVENTOURI T ,BUNACIU C E ,PERDIKATSIS V. Neutron powder diffraction studies of silicon-substitutedhydroxyapatite [J]. Biomaterials,2003,24(23):4205-4211.
(責任编辑:郁慧,顾浩然)
猜你喜欢
现代商贸工业(2017年2期)2017-03-28
江苏农业科学(2016年11期)2017-03-21
科教导刊·电子版(2016年24期)2016-10-29
湖南大学学报·自然科学版(2016年6期)2016-07-14
企业文化·中旬刊(2016年6期)2016-06-16
科技视界(2016年2期)2016-03-30
湖北农业科学(2014年18期)2014-11-20
现代电子技术(2014年8期)2014-09-27
