
以先天性甲状腺功能减退症及矮小为首发症状的假性甲状旁腺功能减退症1例报道并文献复习
2023-06-28 07:10孟令哲初国铭张丹辛颖
中国医科大学学报 2023年6期
孟令哲,初国铭,张丹,辛颖
(中国医科大学附属盛京医院 1.小儿内分泌科;2.遗传科,沈阳 110004)
假性甲状旁腺功能减退症(pseudohypoparathyroidism,PHP)是由于分子缺陷致G蛋白(Gsα)α-亚基与受体结合异常,腺苷酸环化酶活化减弱,细胞内信号传导异常,从而引起靶细胞对促甲状旁腺激素抵抗,部分还可合并对其他激素(如甲状腺激素、促生长激素释放激素、促性腺激素等)的抵抗,造成Albright遗传性骨营养不良症(Albright hereditary osteodystrophy,AHO)畸形、智力异常等[1]。本研究报道了1例先天性甲状腺功能减退症及矮小为首发症状的PHP1a型患儿,分析其临床及遗传学资料,并复习相关文献,以期为PHP的临床诊疗提供参考。
1 材料与方法
1.1 临床资料
患儿,女,6岁,因“确诊先天性甲状腺功能减退症6年,生长缓慢1年”,至中国医科大学附属盛京医院小儿内分泌科就诊。患儿6年前新生儿筛查促甲状腺激素(thyroid stimulating hormone,TSH)异常,后外院诊断先天性甲状腺功能减退症,正规服用左甲状腺素钠治疗控制病情,规律复查甲状腺功能维持良好。家属自觉患儿既往偏矮,具体身高增长情况不详,未特殊诊治。1年前患儿开始出现身高增长缓慢,1年仅增长3 cm。患儿平素食量正常,不挑食,无特殊零食及饮料摄入,运动量少。家属述患儿学习能力尚可,智力正常,沟通能力正常。患儿既往无其他手术外伤史,无传染病接触史。患儿系第1胎第1产,母孕史、出生史无异常,出生体质量3.2 kg,出生身长50 cm。否认父母近亲婚配史,父亲身高172 cm,母亲身高146 cm,否认佝偻病家族史,否认糖尿病、高血脂及高血压等家族病史。否认食物及药物过敏史。
入院体格检查:体温36.1 ℃,脉搏88次/min,呼吸20次/min,血压96/59 mmHg,身高111 cm(位于同年龄同性别第3百分位),体质量23 kg(位于同年龄同性别第3~10百分位),体质量指数(body mass index,BMI)18.67 kg/m2。神清状可,体型偏胖,圆脸,五官正常,皮肤无粗糙,无颈璞,双侧乳房TannerⅠ期,无腋毛,心、肺、腹部查体未见明显异常,双手第4、5掌骨短,双足第4、5跖骨短。外生殖器幼稚型,四肢末梢温,活动自如,神经系统查体未见异常。部分临床特征见图1,患儿双手及母亲双手对比见图2。

图1 患儿的部分临床特征
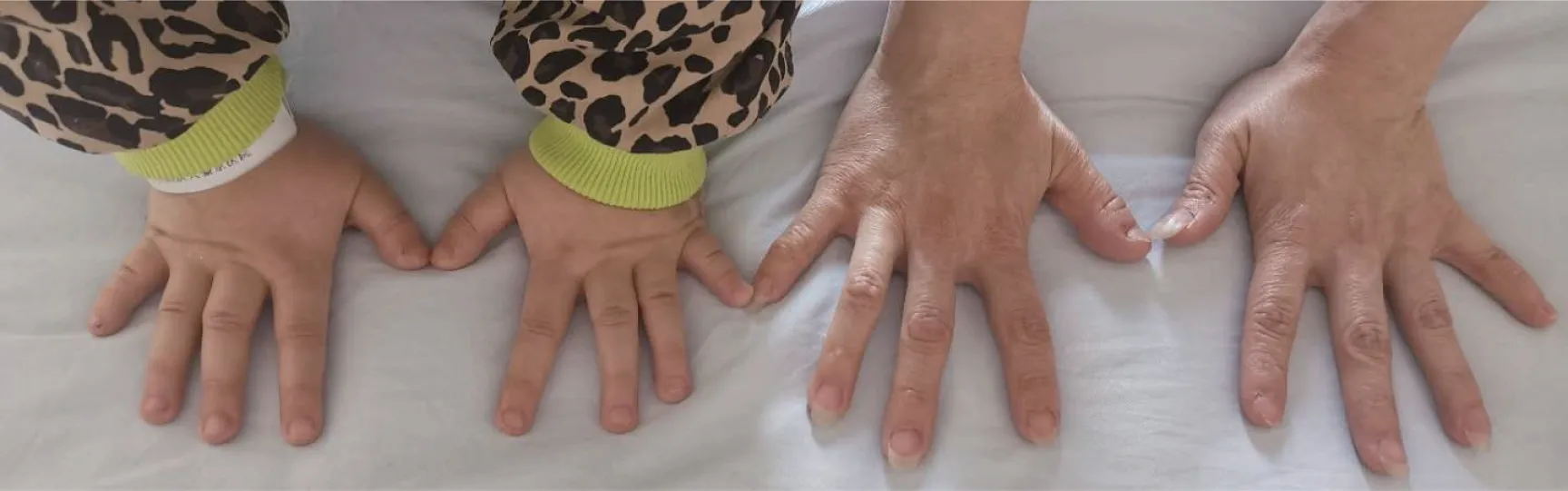
图2 患儿及母亲双手对比图
1.2 检查结果
入院实验室检查:血钙 2.04(2.2~2.7)mmol/L,血磷 2.52(1.2~1.9)mmol/L,甲状旁腺激素(parathyroid hormone,PTH)499.5(8.0~12.0)pg/mL,24 h尿钙0.1(2.5~7.5)mmol/d,24 h尿磷 12.95(23.0~48.0)mmol/d,胰岛素样生长因子130(50~410)ng/mL。左手正位X线片:组成诸骨骨质密度不均匀减低,骨小梁稀疏,掌骨及指骨略短缩,部分略弯曲,钩骨可见囊片状低密度影,指间关节间隙变窄,RUS法计算骨龄9岁,见图3。头CT:双侧顶叶、基底节区见对称性片状高密度影,考虑钙化灶可能性大,其余结构未见明显异常。

图3 患儿左手正位X线片
入院后复查甲状腺功能各指标均在正常范围内,血常规、促肾上腺皮质激素、皮质醇、性激素、人绒毛膜促性腺激素、肝肾功能、心肌酶、血清离子钾钠氯检测,以及心电图、胸部正侧位片、腹腔超声、双肾输尿管超声、甲状旁腺超声、心脏超声、子宫附件超声及垂体增强磁共振成像检查均未见明显异常。染色体呈46,XX。应用精氨酸及左旋多巴完善生长激素(growth hormone,GH)激发试验,测定0、30、60、90、120 min生长激素提示生长激素缺乏,见表1。
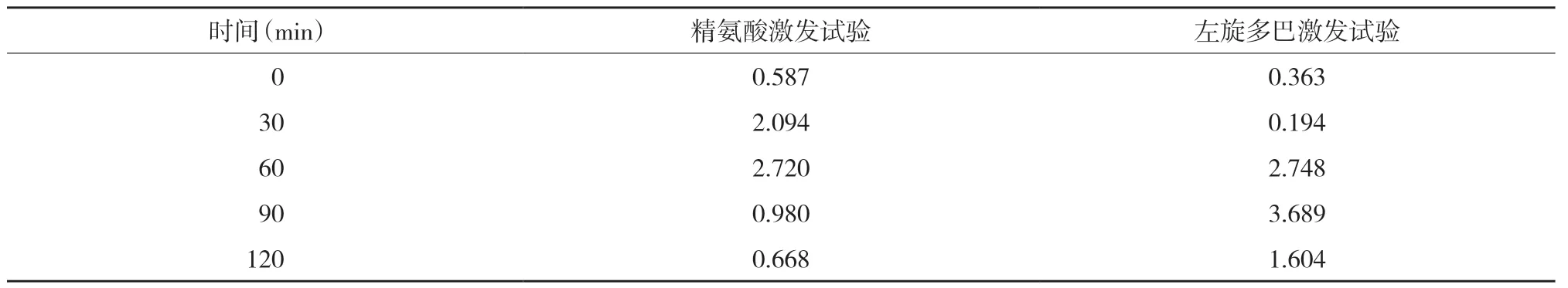
表1 生长激素检测结果(ng/mL)
1.3 遗传学分析
结合患儿临床症状、查体及实验室检查后,考虑诊断PHP1a型。在获得知情同意后,抽取患儿及父母静脉血2 mL,完善基因检测。按照基因组DNA提取试剂盒(Blood DNA Mini Kit,美国QIAGEN公司)说明书提取DNA,应用超微量分光光度仪(Nanodrop 2000,美国Thermo Fisher Scientific公司)行DNA质检。合格样品采用超声打断仪(Covaris S220,美国Thermo Fisher Scientific公司)将其打断至100~700 bp片段,应用标准文库构建试剂盒(北京迈基诺基因科技有限公司)进行PCR扩增。经过生物素标记的探针(北京迈基诺基因科技有限公司)与文库 DNA 进行杂交,用链霉亲和素修饰磁珠共价结合生物素标记的探针,抓取目的基因。经过质控评估后利用NextSeq 500平台(德国Illumina公司)完成二代测序分析。利用Cutadapt程序(https://cutadapt.readthedocs.io/en/stable/)[2]过滤掉原始数据中低质量的读取和接头序列,利用SolexaQA和BWA软件将预处理后数据与人基因组进行比对[3],用GATK程序(https://software.broadinstitute.org/gatk/)对单核苷酸多态性(single nucleotide polymorphisms,SNPs)和插入/缺失(InDels)进行鉴定,再用ANNOVAR程序(http://annovar.openbioinformatics.org/en/latest/)[4]对识别出的SNPs和InDels进行注释。最后用MagicViewer软件(http://bioinformatics.zj.cn/magicviewer/index.php)查看并验证候选SNPs和InDels。应用4种不同生物信息分析工具Ployphen(http://genetics.bwh.harvard.edu/pph2/)、Sorting Intolerant from Tolerant(SIFT,http://provean.jcvi.org/index.php)、PANTHER(http://www.pantherdb.org)和Pmut(http://mmb.pcb.ub.es/PMut/)对所获突变进行致病性分析和预测。通过人类基因突变数据库(http://www.biobase-international.com/product/hgmd)支持结果的新颖性,变异点严格按照美国医学遗传学和基因组学学院(American College of Medical Genetics and Genomics,ACMG)指南进行致病性分析。
2 结果
遗传学分析结果显示,患儿及母亲携带c.92_94delAGA(P.31_32delQKinsQ)杂合突变,见图4。该突变为GNAS基因转录外显子1号外显子整码突变,导致缺失31和32号谷氨酰胺及赖氨酸,插入谷氨酰胺,最终结果为缺失赖氨酸。根据ACMG指南,该突变为疑似致病性变异,与LOVD数据库(http://www.lovd.nl/GNAS)进行对比,虽该整码突变为首发,但此位置赖氨酸缺失已有报道[5]与PHP1a型有关,患儿父亲未见该位点突变。本例患儿具有先天性甲状腺功能减退症、AHO畸形、GH缺乏、甲状旁腺功能减退症,并有GNAS基因突变,诊断PHP1a型。二代测序其余结果为无意义突变,故未列举。出院前予患儿碳酸钙D3 600 mg口服,3次/d,骨化三醇胶丸0.25 μg口服,2次/d,基因结果回报前暂未应用治疗,定期门诊复查。
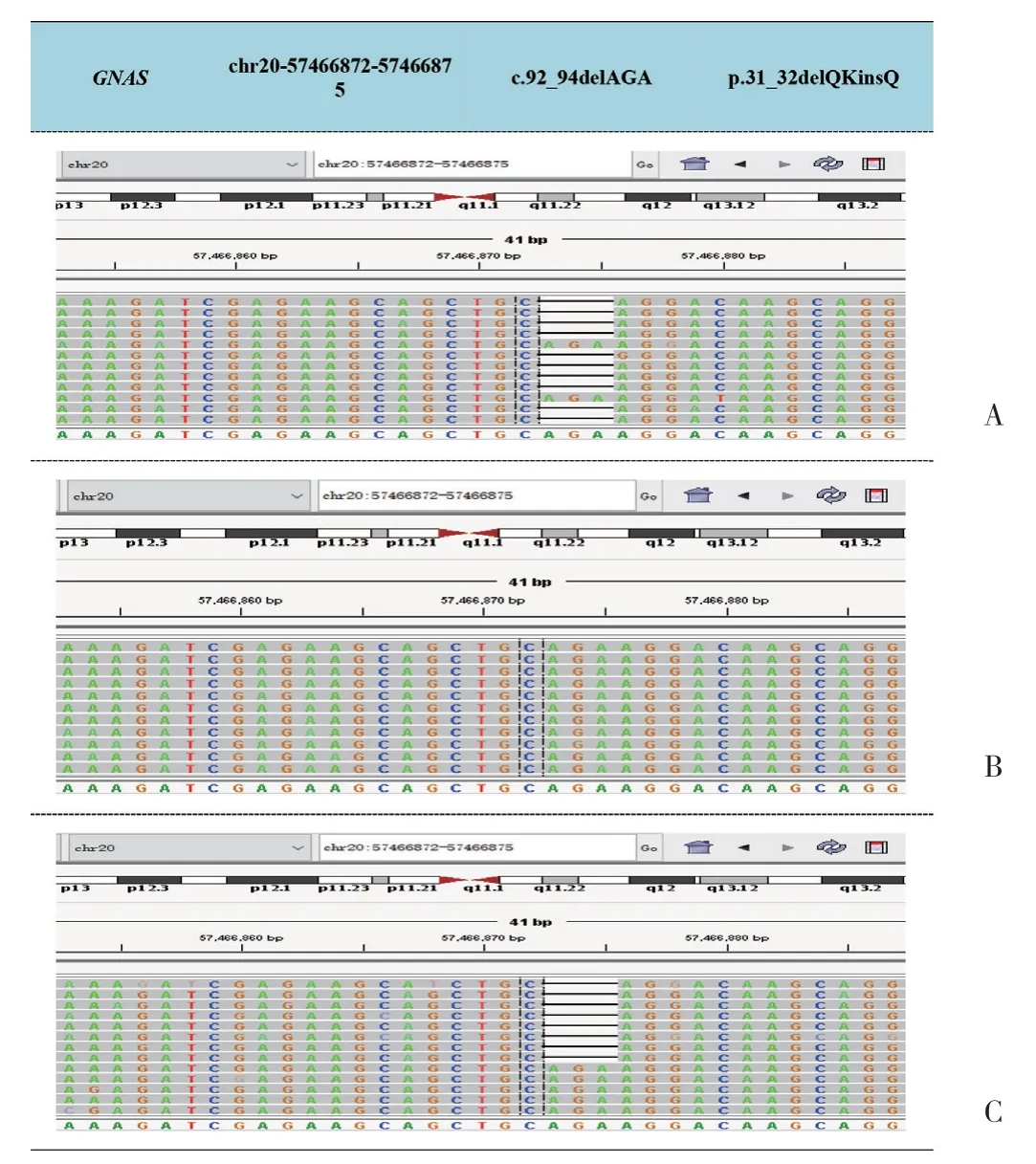
图4 患儿二代测序基因结果
3 讨论
PHP是指由于机体靶组织对PTH产生抵抗所致,具有与甲状旁腺功能减退症相同的生化异常(即低血钾、高血磷)的一组疾病,最早是由ALBRIGHT等[6]首次报道。目前的研究[7]显示,90%的PHP可发现分子学证据。PHP1a型最早被定义为对多种激素(包括甲状旁腺激素及TSH)抵抗、具有AHO特征改变及与Gsα活性减低相关联的疾病[8]。AHO的主要特征包括圆脸、矮胖体态、脊柱畸形和异位钙化。身材矮小是由于骨骺过早闭合,骨骼生长发育期变短所致。骨骼短小可发生于所有骨骼,但以肢端(即手和脚)短小最为明显。
目前认为PHP是由6号染色体上的GNAS基因突变造成,遗传方式为显性遗传或新发突变。本例患儿存在GNAS1号外显子整码缺失突变,造成第32号氨基酸缺失赖氨酸,导致Gsα蛋白活性丧失,TSH、PTH及促生长激素抵抗。临床实验室检查可发现甲状腺功能减退症、PHP1a及生长激素缺乏。PHP1a型患者在新生儿期即可发现TSH抵抗,主要表现为血TSH升高,游离甲状腺素T4(free thyroxine T4,FT4)正常或轻度降低,可被误诊为先天性甲状腺功能减退症[9],国内已有文献[10]报道1例以先天性甲状腺功能减退症为首发症状的PHP。陈晓宇等[11]对70例确诊先天性甲状腺功能减退症患儿进行基因二代测序,发现3例(4%)存在GNAS基因错义突变,TSH均>100 μIU/mL,其中2例患儿FT4明显低于正常值,1例接近正常值下限。PTH抵抗在幼儿期以后才出现,并逐渐出现甲状旁腺功能减退症样表现,同样,短指表现至青春期前逐渐明显[12]。本病例即在新生儿期诊断为先天性甲状腺功能减退症,后逐渐因生长缓慢、手指畸形来诊并确诊。
PHP1a还可能有其他症状,如出生身长较正常新生儿短,发生在其他内分泌系统及生化检查异常之前的肥胖[9]。还有研究[13]发现,PHP1a除了以神经系统双侧基底节钙化为主的异位钙化以外,还可能发生结缔组织异位骨化,有异位骨化发生时不能排除PHP1a。虽然认知障碍与PHP1a有关,但30%的PHP1a患者认知发育正常[14]。除上述症状外,Gsα蛋白活性丧失还可能造成降钙素、促黄体生成素及促卵泡生成素抵抗,导致这些激素水平升高。
临床高度怀疑PHP的患者最终需通过分子诊断确诊。如前所述,PHP1a型主要是由于GNAS基因位点突变所致,突变类型可以是母系遗传也可为新发。1代测序及2代测序均可发现基因点突变,而基因重组突变或基因甲基化缺陷需通过多重连接探针扩增技术发现[15]。尚无证据表明GNAS突变的类型与疾病发病时间、内分泌激素抵抗的严重程度、神经认知表型或AHO特征的数量之间具有相关性。
目前,对PHP以基于患者症状及实验室检查异常的治疗为主。甲状腺旁腺激素抵抗需补充活性维生素D或类似物及钙剂,以抑制PTH水平过高及低钙血症、高磷血症。前者会导致骨骺过早钙化,后者则可致肾及颅内钙沉积。TSH抵抗治疗原则与甲状腺功能减退症或亚临床甲状腺功能减退症治疗原则相同,但是对甲状腺功能减退症的及时诊断与治疗似乎不能阻止PHP1a型患者运动或认知障碍的发展。几乎所有的PHP1a型患者均有身材矮小症状,因此,对于身材矮小合并骨骼畸形的患者,应额外注意PHP的可能。PHP1a所导致的肥胖及其他激素的抵抗也应长期关注随访。
PHP1a型患者从出生时即可发生甲状腺功能减退症,之后出现身材矮小,假性甲状旁腺功能亢进所致骨骺过早闭合、异位骨化及异位钙沉积所致惊厥发作,骨骼畸形,活动受限,认知障碍和精神异常。及早发现和治疗可减轻或延后相关并发症的进展。
猜你喜欢
中国民间疗法(2021年13期)2021-08-30
中老年保健(2021年5期)2021-08-24
小雪花·成长指南(2021年2期)2021-05-20
天津医科大学学报(2021年1期)2021-01-26
中国生殖健康(2020年6期)2020-02-01
天津医科大学学报(2019年3期)2019-08-13
初中生世界·九年级(2019年4期)2019-05-05
中国生殖健康(2018年6期)2018-11-06
中国医疗美容(2015年4期)2015-04-27
山西卫生健康职业学院学报(2014年5期)2014-04-09
