
甲基嘧啶磷原药中残留溶剂气相色谱分析
2023-06-27 12:06王胜得曹金艳杜升华
农药科学与管理 2023年5期
吴 曼,王胜得,曹金艳,陈 明,杜升华
(1.湖南化工研究院有限公司 国家农药创制工程技术研究中心,湖南 长沙 410014;2.农用化学品湖南省重点实验室,湖南 长沙 410014)
农药产品中的杂质情况,是判定农药产品质量的重要指标。杂质不仅会影响农药产品质量和使用效果,还可能会对植物、人和动物、环境以及农产品安全产生重要影响[1]。杂质作为农药的一项关键质量属性,一般包括相关杂质和含量大于0.1%的普通杂质[2]。杂质中的残留有害溶剂,因沸点低、易挥发、毒性大,需要对其残留水平进行严格控制。本文以甲基嘧啶磷原药中的常见残留有机溶剂为例,建立了可与相关杂质同步测定的甲醇、甲基异丁基甲酮(以下简称为MIBK)和甲苯的定量检测方法,旨在对农药原料药中残留溶剂的监测与控制提供借鉴意义。
本文参考农药中有害溶剂测定常见的气相色谱法[3,4],考虑同时测定甲基嘧啶磷原药中其他相关杂质和残留溶剂的实际工作需求,建立了加相对校正因子的气相色谱内标法,既可同时测定甲基嘧啶磷原药中微量残留溶剂甲醇、MIBK和甲苯含量,也无需每次跟随溶剂对照品,可实现简单快速准确的多杂质同步质量控制模式。所述方法对原药中的残留有机溶剂和其他相关杂质均做到了有效分离,以相对保留时间进行溶剂定性,以加相对校正因子的内标法进行定量检测,适用于工厂生产车间的快速品质保障检测。
1 实验部分
1.1 仪器和试剂
1.1.1 仪器 岛津GC-2010 plus型气相色谱仪,配有FID检测器和AOC-20i型自动进样器。
1.1.2 试剂 甲基嘧啶磷标准品(99.0%);4,4’-二甲氧基二苯甲酮标准品(99.0%);甲醇、MIBK、甲苯均为色谱纯(≥99.8%);甲基嘧啶磷原药(≥95.0%)。
1.2 色谱条件 色谱柱:Agilent DB-17,15 m×0.25 mm(id)熔融石英毛细管柱,膜厚0.5 μm;程序升温,初始温度50 ℃,保留2 min,以 20 ℃/min速率升温至240 ℃并保留5 min,再以30 ℃/min速率升温至295 ℃并保留10 min;FID检测器温度:295 ℃;进样口温度:230 ℃;分流比:25∶1;载气:氦气,70 kPa,压力控制模式;进样量:1 μL.
1.3 溶液的配制
1.3.1 内标溶液的配制 准确称取内标(4,4’-二甲氧基二苯甲酮)约0.6 g(精确至0.000 2 g)至50 mL容量瓶中,加45 mL乙腈超声溶解10 min,冷却后用乙腈定容至刻度,摇匀备用(如有沉淀需过滤)。
1.3.2 混合标准储备液的配制 准确称取甲基嘧啶磷标准品、甲醇、MIBK和甲苯标准品各0.06 g(精确至0.000 L2 g)至25 mL容量瓶中,乙腈溶解,定容至刻度,摇匀备用。
1.3.3 混合标准溶液的配制 精密移取标准品储备液0.5 mL至25 mL容量瓶,加入1 mL上述内标溶液,用乙腈稀释至刻度,摇匀备用。
1.3.4 试样溶液的配制 称取约0.6 g(精确至0.000 2 g)甲基嘧啶磷原药样品至25 mL容量瓶中,准确加入上述1 mL内标溶液,用乙腈定容至刻度,摇匀备用。
1.4 样品测定 在上述仪器操作条件下,待仪器稳定后,连续注入数针混合标准溶液,直至相邻2针甲基嘧啶磷(或甲醇、MIBK、甲苯)与内标的峰面积比值相对变化不超过1%后,再按照试样溶液、试样溶液、混合标准溶液、试样溶液、试样溶液、混合标准溶液的进样顺序进行测定。
1.5 计算 残留溶剂的含量Xi(%)按下式进行计算,其中f和RFi值的计算根据其线性方程的斜率进行计算,详见2.3项。
式中:Ai为试样溶液中待测残留溶剂峰面积,Ais为试样溶液中内标物峰面积,C为试样浓度,g/mL,Cis为试样溶液中内标物浓度,g/mL,f为甲基嘧啶磷校正因子,RFi为残留溶剂相对于甲基嘧啶磷的相对校正因子。
2 结果与讨论
2.1 甲基嘧啶磷原药中甲醇、MIBK和甲苯的色谱分析图 甲醇、MIBK和甲苯残留水平较高的甲基嘧啶磷原药典型气相色谱图(图1)。由图可见,甲基嘧啶磷原药中除残留有机溶剂外,还含有多种相关杂质。现行残留溶剂分析方法常见的顶空进样GC法[5],可避免样品本身和样品中的非挥发组分注入气相色谱仪中造成污染。笔者选用了直接进样的气相色谱法,可同时分析甲基嘧啶磷原油中残留有机溶剂和相关杂质(本文只讨论残留溶剂分析结果);并且在对比了多种不同固定液类型毛细管柱和操作条件分离效果的基础上,确定了1.2所述色谱操作条件。该法保证残留溶剂和相关杂质能够在同一色谱条件下全部流出并有效分离,满足了运行一针样品即可完成多组分同步质量控制模式的需求,大大节约了分析时间。

1-甲醇;2-甲基异丁基酮;3-甲苯;4-甲基嘧啶磷;5-内标
2.2 内标物的选择 本研究对比了正二十二烷、邻苯二甲酸二乙酯和4,4’-二甲氧基二苯甲酮等一系列内标物,最终选择4,4’-二甲氧基二苯甲酮(峰5)作为残留溶剂分析的内标物,虽保留时间距离低沸点溶剂较远,但是与图中其他相关杂质完全分离,不与待测组分发生任何化学反应,并且对于同时分析甲基嘧啶磷原油中相关杂质甚至其他高沸点有机溶剂,都是较为理想的内标物。
2.3 线性相关性及校正因子、相对保留时间的测定 线性范围根据实际测定所需浓度范围来确定。精确量取标准品储备液2至10 mL容量瓶中,乙腈稀释,作为标准品稀释液。分别从标准品稀释液中吸取0.1 mL、标准品储备液中吸取0.1、0.5、2.5、12.5 mL至25 mL容量瓶中,加入1 mL上述内标溶液,用乙腈稀释至刻度,配制出在2 ~ 1 250 mg/L浓度范围内系列不同质量浓度的标准溶液,在上述气相色谱分离条件下进行测定。
以标准品与内标物的质量浓度比为横坐标,标准品与内标物的峰面积比为纵坐标,分别测定甲基嘧啶磷、甲醇、MIBK和甲苯的线性方程及其相关系数γ,以其线性方程的斜率计算甲醇、MIBK和甲苯相对于甲基嘧啶磷的相对校正因子RFi,RFi=fi/f甲基嘧啶磷=k甲基嘧啶磷/ki,fi=1/ki,f甲基嘧啶磷=1/k甲基嘧啶磷,其中f甲基嘧啶磷和k甲基嘧啶磷为浓度范围内甲基嘧啶磷的校正因子和线性方程斜率,fi和ki为浓度范围内甲醇(或MIBK、甲苯)的校正因子和线性方程斜率。以各浓度点色谱峰平均保留时间tR计算甲醇、MIBK和甲苯相对于甲基嘧啶磷的相对保留时间,RRT= tR溶剂/tR甲基嘧啶磷。线性关系(图2),相对校正因子等(表1)。

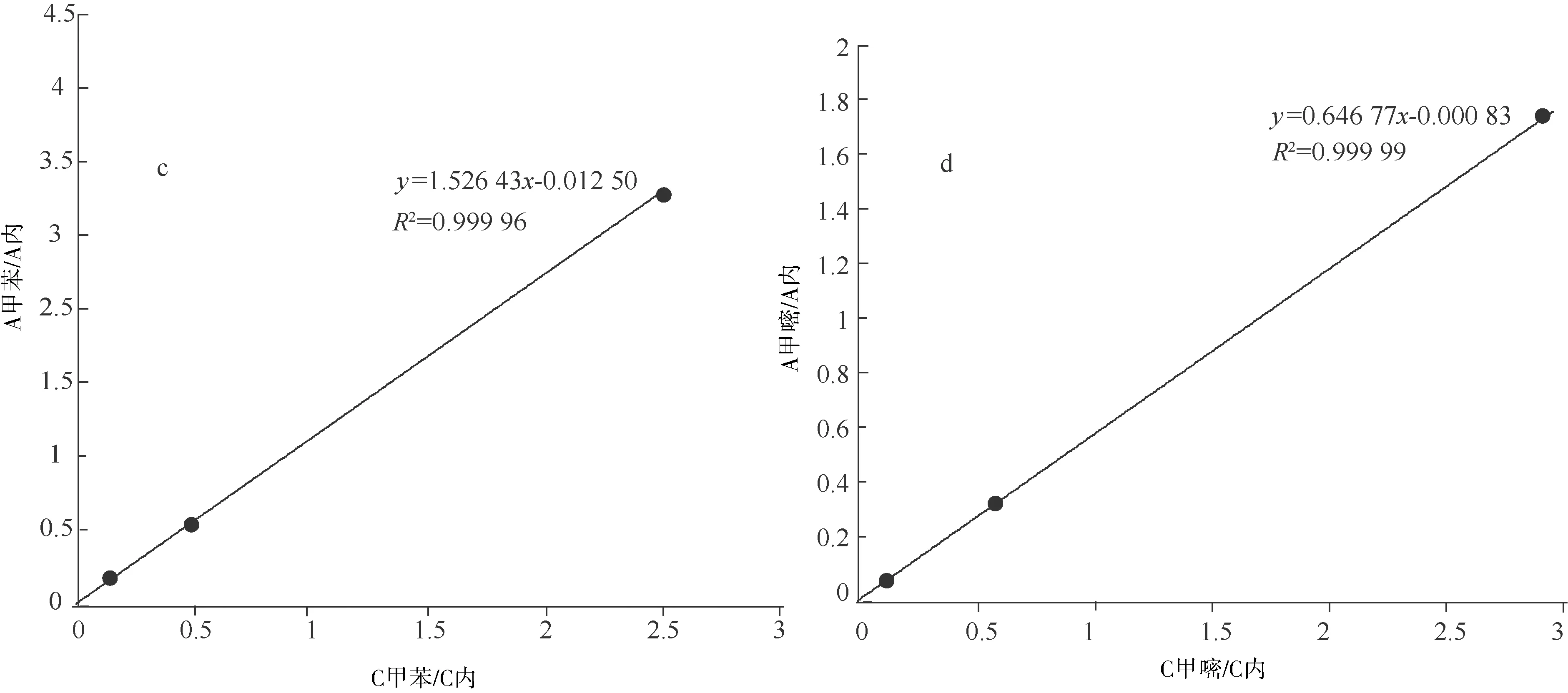
a.甲醇线性关系图;b.MIBK线性关系图;c.甲苯线性关系图;d. 甲基嘧啶磷线性关系图

表1 线性关系和相对校正因子、相对保留时间
2.4 分析方法的精密度试验 在上述气相色谱操作条件下,对同一样品中的甲醇、MIBK和甲苯进行6次重复测定,计算其精密度,结果(表2)。甲醇的标准偏差为0.000 89,相对标准偏差为0.99%,MIBK的标准偏差为0.012,相对标准偏差为0.62%,甲苯的标准偏差为0.000 75,相对标准偏差为1.19%,表明仪器操作条件和分析方法精密度高,重现性好,满足该甲基嘧啶磷原药中残留有机溶剂的定量要求。
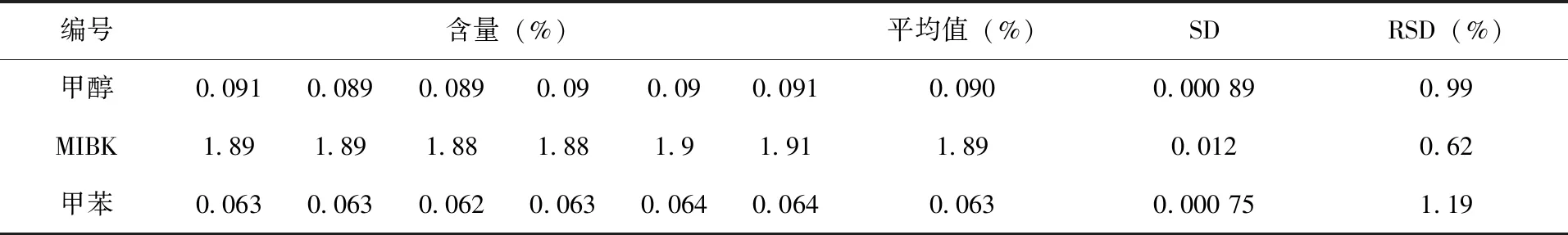
表2 甲醇、MIBK和甲苯精密度测定结果
2.5 分析方法的准确度试验 称取适量的甲基嘧啶磷原药(已测定甲醇、MIBK和甲苯含量),准确加入标准品储备液,制备成5组样品,按照上述气相色谱操作条件测定甲醇、MIBK和甲苯含量,计算回收率,结果(表3)。

表3 回收率测定结果
由表3可知,该方法甲醇、MIBK和甲苯的平均添加回收率为99.07%、100.58%和100.92%,表明仪器操作条件和分析方法可准确测定有效成分含量,满足杂质定量分析要求[6]。
2.6 外标法比较 用外标法和加相对校正因子的内标法同时测定3批不同批次的甲基嘧啶磷原药中残留甲醇、MIBK和甲苯含量。试样按照1.3.4配制进行测定,用加相对校正因子的内标法计算溶剂含量,另取甲醇、MIBK和甲苯对照品溶液进行测定,用外标法计算试样中甲醇、MIBK和甲苯含量,结果(表4)。外标法比对结果表明,加相对校正因子的GC内标法能够准确测定甲基嘧啶磷原药中残留溶剂甲醇、MIBK和甲苯含量。

表4 样品中残留溶剂含量
3 结论
本文建立的加相对校正因子的气相色谱内标法,可在同一色谱条件下同时测定甲基嘧啶磷原药中微量有机溶剂甲醇、甲基异丁基甲酮和甲苯的含量,经线性范围、精密度和准确度的方法验证和外标法比较,表明结果准确,方法简单,可引申为其他残留有机溶剂和相关杂质的定量检测方法,仅需测定其对甲基嘧啶磷的相对校正因子,并定期进行相对校正因子的修正即可,可代替外标法,提高实验效率,对农药原料药研究开发的多杂质同步控制提供了参考意义。
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13
中华养生保健(2020年9期)2021-01-18
无机化学学报(2019年2期)2019-02-27
武警医学(2018年10期)2018-11-06
中国农资(2016年31期)2016-09-13
中国农资(2016年34期)2016-08-11
中国农资(2016年36期)2016-08-09
中国农资(2016年33期)2016-08-02
当代化工研究(2016年6期)2016-03-20
应用化工(2014年7期)2014-08-09
