
Comprehensive and deep profiling of the plasma proteome with protein corona on zeolite NaY
2023-06-26 02:55CongongYnweiLiJieLiLeiSongLingyuChenZhoXuepingLiNingChenLixiLongJinZhoXinHouLiRenXuoYun
Congong M ,Ynwei Li ,Jie Li ,Lei Song ,Lingyu Chen ,N Zho ,Xueping Li ,Ning Chen ,Lixi Long ,Jin Zho ,*,Xin Hou ,Li Ren ,Xuo Yun
a Tianjin Key Laboratory of Composite and Functional Materials,School of Materials Science and Engineering,Tianjin University,Tianjin,300350,China
b Department of Integrative Oncology,Tianjin Medical University Cancer Institute and Hospital and Key Laboratory of Cancer Prevention and Therapy,Tianjin,300060,China
c Department of Proteomics,Tianjin Key Laboratory of Clinical Multi-omics,Tianjin,300308,China
d Department of Clinical Laboratory,Tianjin Medical University Cancer Institute and Hospital,National Clinical Research Center for Cancer,Key Laboratory of Cancer Prevention and Therapy,Tianjin's Clinical Research Center for Cancer,Tianjin,300060,China
Keywords:
NaY
Plasma proteomics
Protein corona
Low-abundance proteins
ABSTRACT
Proteomic characterization of plasma is critical for the development of novel pharmacodynamic biomarkers.However,the vast dynamic range renders the profiling of proteomes extremely challenging.Here,we synthesized zeolite NaY and developed a simple and rapid method to achieve comprehensive and deep profiling of the plasma proteome using the plasma protein corona formed on zeolite NaY.Specifically,zeolite NaY and plasma were co-incubated to form plasma protein corona on zeolite NaY(NaY-PPC),followed by conventional protein identification using liquid chromatography-tandem mass spectrometry.NaY was able to significantly enhance the detection of low-abundance plasma proteins,minimizing the“masking”effect caused by high-abundance proteins.The relative abundance of middleand low-abundance proteins increased substantially from 2.54% to 54.41%,and the top 20 highabundance proteins decreased from 83.63% to 25.77%.Notably,our method can quantify approximately 4000 plasma proteins with sensitivity up to pg/mL,compared to only about 600 proteins identified from untreated plasma samples.A pilot study based on plasma samples from 30 lung adenocarcinoma patients and 15 healthy subjects demonstrated that our method could successfully distinguish between healthy and disease states.In summary,this work provides an advantageous tool for the exploration of plasma proteomics and its translational applications.
1.Introduction
Plasma communicates directly with almost all organs and tissues and its protein composition is differentially regulated during the disease development process.As such,plasma is considered an important source of potential pharmacodynamic biomarkers [1].A comprehensive understanding of plasma proteins in physiological and pathological states can effectively contribute to the discovery of molecular markers and drug targets.However,the vast dynamic range of plasma,estimated at 12-13 orders of magnitude [2],renders plasma proteome analysis based on mass spectrometry (MS)extremely challenging.About 22 proteins with concentrations up to mg/mL in plasma account for 99% of the total proteins,and 90% of them are albumin,immunoglobulin,and fibrinogen[2,3].Thousands of other proteins of interest,such as tissue leak proteins and signaling molecules [4],exist in plasma at concentrations as low as ng/mL or even pg/mL[2].While these low-abundance proteins may be important indicators of disease,the overwhelming “masking”effect caused by more abundant functional proteins makes detecting them quite difficult[5],even by the most advanced MS techniques.Therefore,it is imperative to develop robust proteomics technologies to achieve comprehensive and deep profiling of the plasma proteome,particularly for low-abundance proteins.
To improve the coverage of the plasma proteome,immunoaffinity-based approaches have been developed to deplete 6-20 high-abundance proteins from plasma[6-8]and analyze the remaining proteins by liquid chromatography-tandem mass spectrometry (LC-MS/MS).This approach is able to increase the identification of plasma proteins to 500-800,but high-abundance proteins may form complexes with low-abundance proteins of interest[9],and discarding these carrier proteins inevitably results in the loss of their valuable cargo [10,11].Alternatively,when combined with the removal of high-abundance proteins,peptide fractionation enables the stable quantitative detection of thousands of proteins [7,12],but at the cost of severe sacrifices in analysis time and throughput.
Recently,protein corona has exhibited great promise in plasma proteomics profiling.Particles exposed to biological fluids such as plasma will adsorb proteins,forming a protein layer known as“protein corona” [13-15].Protein corona can act as a “concentrator”of those serum/plasma proteins with affinity for the particle surface [16-20],thus improving the detection of low-abundance proteins [21,22].Blume et al.[19] developed a plasma protein detection platform based on multiple magnetic nanoparticles and the protein corona strategy,which detected approximately 2000 proteins in 141 subjects.The combined use of multiple nanoparticles with complementary properties contributed to the generation of distinct corona patterns and thus improved plasma proteome coverage.Unfortunately,however,for a single plasma sample,the methodology requires separate co-incubation of these nanoparticles with the sample and multiple assays with LC-MS/MS,which inevitably results in an increase in detection time and a decrease in reproducibility.As described in that paper,705 LC-MS/MS assays were required for 141 plasma samples.What's more,noting the huge number of the entire plasma proteome (about 20,000) [23] and the low emergence rate of new biomarkers (less than 2 per year)[24],it is urgent to increase the coverage of plasma proteins by developing new and simple methods,especially those utilizing only one kind of particle.
Zeolite,as an excellent adsorbent,has exhibited outstanding performance in the field of gas and heavy metal ion separation and adsorption[25],and besides,it has attracted great research interest in protein adsorption-related fields because of its unique and abundant surface properties.It was found that with the strong polarity of zeolite cavities,zeolites can interact strongly with protein molecules containing polar groups due to the strong polarity of zeolite cavities[26].Micropores on the zeolite surface significantly affect the protein-zeolite interaction [27].Some residues of amino acids and size-matched secondary structures of proteins may be recognized by the microporous openings,which contribute to the reduction of the adsorption free energy and thus enhance the interaction between protein molecules and porous zeolite.Furthermore,the negative surface charge,surface silica hydroxyl groups,and adjustable hydrophobicity allow zeolites to interact with proteins through various forces such as van der Waals forces,electrostatic interactions [28],and hydrogen bonding [29].These properties combined make zeolites capable of capturing a broader range of proteins through the synergy of multiple forces.Zhang et al.[30] used zeolites to concentrate trace peptides in large solution volumes for matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) analysis,and Rahimi et al.[28] demonstrated that NaY had no significant adsorption effect on acidic pI-valued serum albumin at pH 7.5,which supports that zeolite NaY may have great potential in solving“masking” issues caused by high-abundance proteins during the plasma proteome analysis.
In this work,we synthesized zeolite NaY and proposed a rapid,simple,and reproducible method based on the plasma protein corona that forms on zeolite NaY (termed as NaY-PPC),aiming to achieve a comprehensive characterization and deep profiling of the plasma proteome(Fig.1).Specifically,zeolite NaY and plasma were co-incubated to form NaY-PPC,followed with LC-MS/MS for protein identification.The performance of the methodology was comprehensively evaluated in terms of the number of identifications,qualitative and quantitative reproducibility,changes in the relative abundance of proteins,and detection depth.In addition,the effectiveness of differentiating healthy individuals from cancer patients was tested using a pilot study of 30 lung adenocarcinoma(LUAD) subjects and 15 healthy individuals.
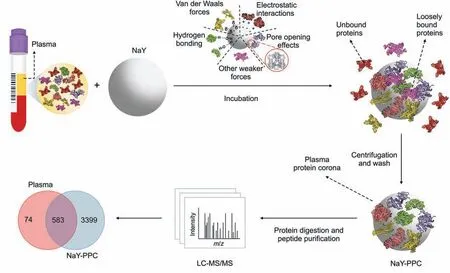
Fig.1.Schematic workflow of the plasma proteomics platform based on NaY-PPC and LC-MS/MS.Our workflow consists of the following steps:1) incubation of NaY with plasma and the formation of protein layers possibly by van der Waals forces,electrostatic interactions,hydrogen bonding,pore opening effects,or other weaker forces;2)centrifugation and washing to obtain a stable plasma protein corona,discarding unbound as well as loosely bound proteins; and 3) digestion of NaY-PPC and purification of peptides for LC/MS-MS analysis.LC-MS/MS:liquid chromatography-tandem mass spectrometry;NaY-PPC:plasma protein corona formed on zeolite NaY;Plasma:human plasma,and pure plasma control for direct LC-MS/MS detection.
2.Materials and methods
2.1.Reagents and chemicals
Ethylenediaminetetraacetic acid(EDTA),chloracetamide,tris(2-carboxyethyl) phosphine hydrochloride (TCEP),and fluorescein isothiocyanate (FITC) were obtained from Sigma-Aldrich (Saint Louis,MO,USA).Coomassie brilliant blue R-250,tris(hydroxymethyl)aminomethane-hydrochloride (Tris-HCl),and sodium deoxycholate were obtained from Solarbio(Beijing,China).Formic acid (FA),acetonitrile (ACN),and High-Select™Top 14 Abundant Protein Depletion Resin (A36370) were purchased from Thermo Fisher Scientific (Waltham,MA,USA).Sodium hydroxide (NaOH),sodium aluminate (NaAlO2),and silica solution were respectively purchased from Tianjin Jiangtian Chemical Technology Company Limited (Tianjin,China),Shanghai Aladdin Bio-Chem Technology Company Limited (Shanghai,China),and Qingdao Ocean Chemical Company Limited (Qingdao,China).Potassium bromide (KBr) was purchased from Heowns Biochem Technologies Company Limited(Tianjin,China).Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample loading buffer (5X) was bought from Beyotime (Shanghai,China).iRT standards were purchased from Biognosys (Schlieren,Switzerland).Trypsin was obtained from Promega (Madison,WI,USA).
2.2.Synthesis and characterization
2.2.1.Synthesis
Zeolite NaY was synthesized following the below gel formulation: 1 SiO2:0.09 Al2O3:0.37 Na2O:16 H2O (molar ratio).2.73 g of AlNaO2and 4.15 g of NaOH were added to a polytetrafluoroethylene reactor containing 20 mL of water and stirred to dissolve,after which 44.44 g of silica sol was slowly added and stirred thoroughly until a homogeneous silica-aluminum gel was formed.The hydrothermal reaction was carried out at 120°C for 72 h.Then the filter cake was filtered and washed with distilled water,followed by drying at 100°C for 12 h and calcination in a muffle furnace at 350°C for 2 h to get zeolite NaY.
2.2.2.Characterization
The morphology of zeolite NaY was characterized using a scanning electron microscope (SEM,S-4800,Hitachi Limited,Tokyo,Japan) at an accelerating voltage of 3 kV.Energy dispersive X-ray spectroscopy (EDS) was carried out with an EDAX Octane Elect Super spectrometer (EDAX,Mahwah,NJ,USA)to analyze the elemental composition of NaY.An X-ray diffractometer (D8 Focus,Bruker,Karlsruhe,Germany) operating at 40 kV and 40 mA was used to analyze the X-ray diffraction(XRD)patterns of NaY.Flourier transform infrared spectroscopy (FTIR) was detected on a Thermo Fisher Nicolet IS10 FT-IR spectrometer with Zeolite NaY pellets compressed in KBr.Dynamic light scattering (DLS),zeta potential,and confocal laser scanning microscopy (CLSM) characterization are detailed in the Supplementary data.
2.3.Collection of plasma samples
This research was granted by the Ethics Committee of Tianjin Medical University Cancer Institute and Hospital (Ethical batch number: E2020610),and was conducted in strict adherence to the principles of the Declaration of Helsinki.In addition,written informed consent was obtained from all subjects.
2.4.NaY-PPC preparation and digestion
20 μL of zeolite NaY dispersion(25 mg/mL)and 20 μL of plasma were added to 160 μL of Tris-EDTA solution with a pH of 8.0.NaY/plasma mixture was incubated for 30 min at room temperature,and then centrifuged at 12,000gfor 5 min,and the supernatant was discarded.The precipitate was washed three times with 500 μL Tris-EDTA to obtain NaY-PPC.
To digest NaY-PPC,50 μL of Tris-HCl buffer with TCEP and chloroacetamide was added to each sample and shaken at 95°C for 10 min in a metal incubator.After that,1 μg of trypsin was added to the sample and incubated at 37°C for 3 h.The digestion process was terminated with 2% FA,followed by centrifugation and collection of the supernatant.Next,a 200 μL pipette tip equipped with SDB-XC(CDS Empore™2240,CDS Analytical,Oxford,PA,USA)was used to desalinate the collected supernatant.First,the pipette tip was washed with a solution of 0.1% FA and 50% ACN.Then,the sample was loaded after equilibration with 0.1%FA and centrifuged at 1,000gfor 3 min.Next,200 μL of 0.1%FA was added to wash the sample tip,followed by 200 μL of a solution containing 0.1%FA and 50% ACN to elute the desalted peptides.Finally,the peptide eluate was dried with a vacuum and stored at -20°C for subsequent LCMS/MS testing.
2.5.Sample preparation and digestion of the pure plasma control and the depleted plasma
50 μL of Tris-HCl buffer containing TCEP and chloroacetamide was added to the plasma and shaken for 10 min at 95°C in a metal incubator.The sample was then digested,desalted,and dried,following the same procedure as the corresponding procedure for NaY-PPC described above.The dried peptides were obtained for subsequent LC-MS/MS testing.
To obtain the depleted plasma,the commercial High-Select™Top 14 Abundant Protein Depletion Resin was used to remove the 14 most abundant proteins from plasma.Detailed sample preparation and digestion procedures for depleted plasma are shown in the Supplementary data.
2.6.LC-MS/MS analysis and data processing
2.6.1.Data-independentacquisition(DIA)
Dried peptide samples were resuspended in a loading buffer(3%ACN,0.1% FA) with iRT standards,and analyzed on a Thermo Scientific UltiMate 3000 UHPLC system coupled with an Orbitrap Q Exactive HF mass spectrometer.A 90-min separation gradient was established with mobile phase A (2% ACN,0.1% FA) and mobile phase B (0.1% FA in ACN) (0-8 min,3%-6% B; 8-9 min,6% B;9-25 min,6%-12% B; 25-60 min,12%-24% B; 60-70 min,24%-35% B; 70-76 min,35%-90% B; 76-85 min,90% B; 85-86 min,90%-6%B;86-90 min,6%B),where the detection time for the MS was 25-85 min,and therefore also referred to as the 60 min effective gradient.Peptides were ionized at 2 kV.The MS was operated in DIA mode with anm/zscan range of 350 to 1,500 and a resolution of 60,000.The maximum injection time (IT),automatic gain control (AGC) target,and inclusion list were set to 20 ms,1 × 106,and 40 windows,respectively.High energy collisional dissociation(HCD)was employed for precursor ion fragmentation,where the normalized collision energy was 28.The maximum ion injection time of the MS2 was set to automatic and the resolution was 30,000.
The 70 min separation gradient(30 min effective gradient)used for the 10 replicates of the batch-to-batch stability test(Section 3.3)was as follows: 0-3 min,3%-8% B; 3-40 min,8%-40% B;40-45 min,30%-40%B;45-50 min,40%-95%B;50-65 min,95%B;65-66 min,95%-5%B;66-70 min,5%B,where 20-50 min is the MS detection time,and therefore also referred to as the 30 min effective gradient.
The 150 min separation gradient (120 min effective gradient)used for some of the samples in Section 3.3 were the following:0-8 min,3%-6% B; 8-9 min,6% B; 9-30 min,6%-12% B;30-105 min,12%-24%B;105-125 min,24%-35%B;125-126 min,35%-90% B; 126-141 min,90% B; 141-142 min,90%-6% B;142-150 min,6%B,where 17-137 min was the MS detection time,and therefore also referred to as the 120 min effective gradient.
2.6.2.DIArawdataprocessing
Spectronaut software (v14.5.200813.47,784) was utilized to analyze the DIA data and the default settings were used for the software parameters,where the Q cutoff values for proteins and precursors were set to 0.01.Proteomics data were normalized by median correction.The generation of the library for DIA data analysis is detailed in the Supplementary data.
2.6.3.Data-dependentacquisition(DDA)
Dried peptide samples were resuspended in a loading buffer(3%ACN,0.1%FA)with iRTstandards,and analyzed on a Thermo Scientific UltiMate 3000 UHPLC system coupled with an Orbitrap Q Exactive HF mass spectrometer.A 90 min separation gradient was established with mobile phase A(2%ACN,0.1%FA)and mobile phase B(0.1%FA in ACN) (0-8 min,3%-6% B; 8-9 min,6% B; 9-25 min,6%-12% B;25-60 min,12%-24%B;60-70 min,24%-35%B;70-76 min,35%-90%B;76-85 min,90%B;85-86 min,90%-6%B;86-90 min,6%B),where the detection time for the MS is 25-85 min,and therefore also referred to as the 60-min effective gradient.The MS was run at a resolution of 120,000 in them/zrange of 300 to 1500.The maximum IT and AGC were set to 40 ms and 3× 106,respectively.40 peptide parent ions with the strongest ion intensities were selected from the MS pre-scan to continue fragmentation in the secondary MS.
The 150 min separation gradient (120 min effective gradient)used for some of the samples in Section 3.3 were the following:0-8 min,3%-6% B; 8-9 min,6% B; 9-30 min,6%-12% B;30-105 min,12%-24%B;105-125 min,24%-35%B;125-126 min,35%-90% B; 126-141 min,90% B; 141-142 min,90%-6% B;142-150 min,6%B,where 17-137 min was the MS detection time,and therefore also referred to as the 120 min effective gradient.
2.6.4.DDArawdataprocessing
MaxQuant software (2.0.1) was used for DDA data processing with default settings.
2.7.SDS-PAGE electrophoresis
0.5 μL of plasma and NaY-PPC obtained by co-incubation of zeolite NaY with plasma were resuspended with SDS-PAGE loading buffer,respectively,and then boiled at 100°C for 5 min.The plasma sample was loaded directly onto a 5%-12%Bis-Tris gel,and the NaYPPC sample was centrifuged at 12,000gfor 5 min before loading the supernatant onto the gel.The gel was stained with Coomassie brilliant blue R-250 after running at 120 V for approximately 90 min and photographed with a cell phone.
2.8.Statistical analysis
Student'st-test was used for statistical analysis,and*P<0.05 was considered a significant difference.For plasma cohort studies of LUAD patients and healthy subjects,the cohort was corrected with median normalization,and proteins present in over 70%of the samples were chosen for further analysis.Proteins withP<0.05 after correction by the Benjamini-Hochberg method (P-adjust<0.05) and fold change (FC (T/H)) >2 or <0.5 between the tumor group and healthy group were considered as differentially expressed proteins.
3.Results and discussion
3.1.Synthesis and characterization of zeolite NaY
In this work,we prepared zeolite NaY with homogeneous particle size to adsorb proteins from plasma for efficient LC-MS/MS proteomics analysis.The zeolite NaY was characterized by SEM,EDS,XRD,and FTIR.The particle size of NaY was about 400 nm,with a relatively uniform distribution,and the crystals showed a polyhedral shape and a flat and smooth surface (Fig.2A).The EDS showed that NaY consisted mainly of Si,Al,O,and Na(Fig.2B).The XRD results (Fig.2C) showed that 6.2°,10.1°,11.9°,15.7°,18.6°,20.4°,22.5°,23.5°,26.8°,29.3°,30.4°,31.0°,32.0°,33.7°,and 37.6°corresponded to(111),(220),(311),(331),(511),(440),(620),(533),(642),(733),(823),(555),(840),(664),and (666) crystal planes of zeolite NaY,respectively [31,32].The 3,450 cm-1band in the FTIR spectrum (Fig.2D) was attributed to Si-OH stretching vibrations in the zeolite,and the peak around 1,637 cm-1was associated with the bending vibration of the absorbed water.The bands at 1,018 cm-1and 1,137 cm-1belonged to the asymmetric stretching vibrations of the internal tetrahedra and external linkage,respectively,while the bands at 721 cm-1and 788 cm-1corresponded to the symmetric stretching vibrations of the internal tetrahedra and external linkage,respectively [33].The peak at 578 cm-1was associated with the double-ring external linkage peak of the zeolite NaY[31],and the absorption band at 458 cm-1belonged to the bending vibrational absorption peak of the internal tetrahedral T-O (T = Si or Al) of NaY zeolite [34].The above results demonstrate the successful preparation of NaY with relatively uniform particle size distribution and good crystallinity.
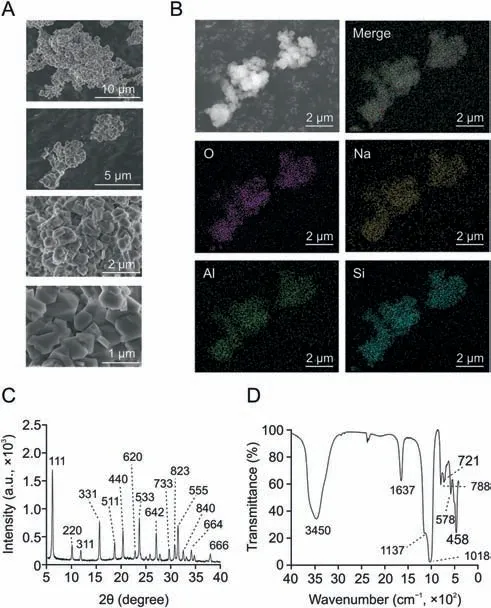
Fig.2.Characterization of zeolite NaY.(A)Scanning electron microscope(SEM) image of the synthesized NaY.(B) Energy dispersive X-ray spectroscopy (EDS) of the synthesized NaY.(C) The X-ray diffraction (XRD) of the synthesized NaY.(D) The Flourier transform infrared (FTIR) spectroscopy of the synthesized NaY.Merge: the image of four elements O,Na,Si,and Al superimposed on each other.
3.2.NaY-PPC-based proteomic analysis strategy
As shown in Fig.1,NaY was first incubated with trace plasma(20 μL) for 30 min at room temperature to form a protein layer.After centrifugation and discarding the supernatant,the NaYprotein complex was washed 3 times with buffer to remove unadsorbed and weakly adsorbed proteins[35],thereby obtaining a stable and repeatable NaY-PPC.Finally,the NaY-PPC underwent reductive alkylation,digestion,and desalting to obtain the peptides,which were then used for LC-MS/MS detection.
DLS,zeta potential,and CLSM were used to characterize NaY before and after co-incubation with plasma,further demonstrating the formation of NaY-PPC(Fig.S1).In particular,a larger size of NaY was observed after incubation with plasma compared to before incubation,and the ζ-potential(-12.0±3.1 mV)was more positive than that of bare NaY (-33.6 ± 1.4 mV) but still negative.Similar phenomena were found in other materials such as nanodiamonds[18] and polystyrene [36].The NaY surface showed green fluorescence after co-incubation with FITC-labeled plasma proteins,demonstrating the protein adsorption phenomenon of NaY.
Next,we counted the number of detected proteins and peptides to initially evaluate the effectiveness of the methodology.Peptides from NaY-PPC were detected in DIA mode in LC-MS/MS with a 60 min effective gradient,and protein identification and quantification were performed using Spectronaut.Pure plasma was used as a control group,and its peptides obtained by direct digestion were analyzed by MS under the same conditions.Figs.3A-C display the number of proteins and peptides from NaYPPC and pure plasma identified by LC-MS/MS.Approximately 4,000 proteins and 31,000 peptides were identified from NaY-PPC,presenting a huge advantage compared to the 600 proteins and 4,000 peptides that were obtained in the pure plasma control(see Supplementary data 1 for a list of all identified proteins).Of these,3399 proteins were uniquely identified by our method.In direct data-independent acquisition (dDIA) mode,about 2,500 proteins and 15,000 peptides were identified in NaY-PPC,while only about 500 proteins and 3,000 peptides were detected in pure plasma(Fig.S2; Supplementary data 1).
Furthermore,we compared the NaY-PPC-based method with a commercial immunoaffinity-based top 14 plasma high-abundance protein depletion kit,with direct digestion of pure plasma remaining as a control.For comparison,mixed plasma samples,the same MS instrument,and gradient(30 min effective gradient,DIA mode)were used,and the MS data were subjected to the same postprocessing.The results showed that the NaY-PPC-based method identified more than 3,100 proteins and 23,000 peptides,with 1.7-and 4.8-fold more proteins and 2.6- and 5.7-fold more peptides compared with the depleted plasma and pure plasma,respectively(Figs.3D-F; Supplementary data 1).Moreover,NaY-PPC effectively covered most of the proteins detected in depleted plasma and pure plasma (Fig.3D).The above results initially demonstrate the powerful capability of NaY-PPC to collect thousands of protein molecules that are undetectable by conventional plasma proteomics.
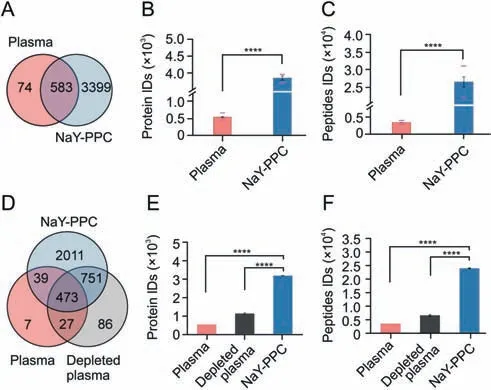
Fig.3.Profiles of proteins and peptides identified from NaY-PPC,depleted plasma,and pure plasma control.(A) Venn diagram depicting the number of unique and common proteins between plasma control and NaY-PPC.Proteins identified in at least one of the three biological replicates of plasma control or NaY-PPC were counted.(B,C) The number of proteins (B) and peptides (C) identified in plasma control or NaY-PPC.The median and standard deviation of the three biological replicates are shown as bar graphs.The lower red dash depicts the number of proteins or peptides detected in all three replicates,and the upper red dash depicts the number of proteins or peptides detected in any of the three biological replicates.(D) Venn diagram depicting the number of unique and common proteins between plasma control,plasma depleted of top 14 high-abundance proteins,and NaY-PPC.(E,F) The number of proteins (E) and peptides (F) identified in plasma control,plasma depleted of top14 high-abundance proteins,and NaY-PPC.The median and standard deviation of the three biological replicates are shown as bar graphs.Plasma: pure plasma control for direct liquid chromatography-tandem mass spectrometry(LC-MS/MS)detection;Depleted plasma:plasma depleted of top 14 high-abundance proteins; NaY-PPC: plasma protein corona formed on zeolite NaY.****P <0.0001.
In addition,the NaY-PPC-based method for plasma proteomics analysis has many advantages,such as low cost,low sample input requirement,and rapid and simple.This is in contrast to the high costs,high-volume sample,time-consuming,and complex workflows of traditional workflows[37,38].Specifically,zeolite NaY acts as an excellent adsorption material,having been widely used in the separation and adsorption of gases and heavy metal ions,and also being cheap and easy to obtain.The low sample input volume makes it possible to extend the assay from venous blood to fingertip blood,which has clinical appeal in many cases,particularly when testing plasma from infants and elderly patients [39].It takes only about 6 h to go from plasma proteins to peptides when using our method,and multiple samples can be prepared simultaneously.More importantly,the whole sample preparation process is extremely simple.For one thing,no pre-fractionation and highabundance plasma protein depletion steps are required; for another,only one type of particle,NaY,is needed,reducing many repetitive processing steps in incubation,washing,and detection compared to the combined use of multiple particles.
3.3.Reproducibility and stability of qualitative and quantitative performances
Excellent reproducibility and stability are among the key requirements for quantitative proteomics applications.The intraand inter-batch reproducibility and stability of the method were evaluated with three biological replicates from the same batch and 10 replicates prepared from different batches,respectively.Besides,to save testing time,the 10 replicates from different batches were tested using a 30 min effective gradient of DIA(Supplementary data 2).As shown in Fig.4A,about 94% of the corona proteins are common across the three biological replicates.The coefficient of variation (CV) of the proteins detected in NaYPPC showed a median CV of 11.58% (Fig.4B) and a mean value of 17.43%.The median CV of pure plasma was lower (9.35%) due to the fact that most of the proteins identified in pure plasma are high- to middle-abundance proteins that are easily detectable.Furthermore,the Pearson correlation coefficient was >0.9 for both 3 replicates within a batch(Fig.4C)and the 10 replicates between batches(Fig.4D),proving that our data are reproducible and solid,which is largely attributed to the simple workflow of this NaYPPC-based method.
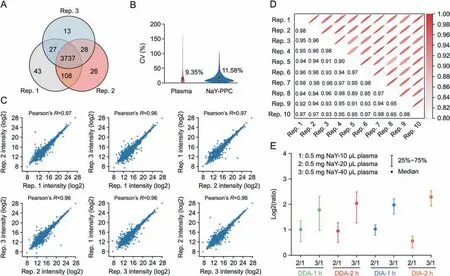
Fig.4.Reproducibility and stability of qualitative and quantitative performances.(A)Venn diagram of the proteins identified in the three biological replicates of NaY-PPC.(B)CV of plasma control and NaY-PPC.Inner boxplots report 25% (lower hinge),50%,and 75% quantiles (upper hinge).Whiskers indicate observations equal to or outside hinge ±1.5 ×interquartile range (IQR).Outliers (beyond 1.5 × IQR) are not plotted.(C) Person correlation analysis of three biological repeats of NaY-PPC.(D) Pearson correlation analysis of 10 inter-batch replicates of NaY-PPC.(E) The ratio of protein intensity for 3 groups with different plasma additions.Ratio = Protein intensity of group m/Protein intensity of group 1(0.5 mg NaY-10 μL plasma).Plasma: pure plasma control for direct liquid chromatography-tandem mass spectrometry (LC-MS/MS) detection; NaY-PPC: plasma protein corona formed on zeolite NaY; CV: coefficient of variation; DDA: data-dependent acquisition; DIA: data-independent acquisition; Rep.n: the nth repetition.
To test the quantitative accuracy of the method,0.5 mg NaY was co-incubated with 10,20,and 40 μL plasma,respectively.Proteins adsorbed by NaY were detected using the 60 min effective gradient and 120 min gradient in DDA and DIA modes,with DDA files processed with Maxquant and DIA processed with Spectronaut(Supplementary data 3).We calculated the ratio of protein intensities of the co-identified proteins for the 3 groups under the same test conditions.Fig.4E showed that the quantitative protein intensities were consistent with the plasma addition.The 20 μL and 40 μL groups showed a 2- and 4-fold relationship in protein intensity compared to the 10 μL plasma group,respectively.These results suggest that our method can accurately reflect the changes in protein abundance,which is critical for biomarker screening.
3.4.Signal attenuation of high-abundance proteins and signal amplification of low-abundance proteins
Deep plasma proteome analysis has always been a challenge for traditional methods.Human plasma has a vast dynamic range,and a few highly abundant proteins produce large amounts of abundant peptides after enzymatic digestion,which dominate the MS signal[40].Moreover,leaked or secreted proteins from the tumor microenvironment are heavily diluted upon entering the circulation [2,4],resulting in their extremely low concentrations in the blood,and current proteomics techniques are still unable to reach the required detection depth to reveal the information of these proteins.Next,we further evaluated the performance of our method in addressing the “masking” effect caused by highabundance proteins,as well as the detection of low-abundance proteins in complex mixtures.
Fig.5A exhibits the results of SDS-PAGE of plasma control and NaY-PPC.The protein molecular weight distribution pattern indicated that a variety of different components together make up the NaY-PPC.The intensity of the bands of albumin(molecular weight~66.5 kDa) was substantially weakened in the NaY-PPC,and multiple bands were clearly visible even near the albumin bands.In contrast,the overwhelming signal of albumin in the plasma control severely masked other proteins in its vicinity (55-70 kDa).These findings suggest that NaY can effectively eliminate the masking of albumin,the most abundant protein in plasma.

Fig.5.Changes in the relative abundance of high- and low-abundance proteins before and after NaY adsorption.(A) Results of sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of plasma control and NaY-PPC.(B) Heat map of the 20 most abundant proteins in plasma (left) and NaY-PPC (right).Protein rows are ranked according to the mean RPA values (from highest to lowest) in plasma (left panel) or NaY-PPC (right panel).(C) Proportions of proteins with different abundance intervals in plasma control and NaY-PPC,respectively.The proteins in the plasma control were sorted in descending order by mean RPA,and the top 100 proteins were divided into five intervals,and then the proportions of these proteins in the plasma control (left) and NaY-PPC (right) were calculated separately.“n-m” denotes the proteins ranked nth to mth in descending order of relative abundance in the plasma control.“Others” refers to proteins in the sample other than these 100 top proteins.Marker: protein ladder to indicate the molecular weight of a protein band; Plasma: pure plasma control; NaY-PPC: plasma protein corona formed on zeolite NaY; RPA: relative protein abundance.
Next,a comprehensive comparison of the proteins identified in NaY-PPC and pure plasma control was performed.To further analyze the protein composition,the relative protein abundance(RPA) of each protein in the three biological replicates of the NaYPPC group or plasma control group was first calculated,which in turn led to the mean RPA of each protein in the NaY-PPC group or plasma control group (Supplementary data 4).The top 20 most abundant proteins identified in plasma,as well as NaY-PPC,are shown as heat maps (Fig.5B; Tables S1 and S2).Notably,several interesting facts are revealed here.The proteins that are richer in plasma are not the major proteins in the protein corona,and these proteins have markedly lower RPA in NaY-PPC in comparison with plasma control.For example,the RPA of albumin decreased from 37.30% in pure plasma to 0.73% in NaY-PPC,and transferrin decreased from 2.80% to 0.05%.The relative abundance order of albumin decreased from 1st in pure plasma to 21st in NaY-PPC,and transferrin decreased from 7th to 168th.(Table S1).Reversely,the RPA of some low-abundance or even non-directly detectable proteins was significantly increased by the adsorption of NaY.For instance,the RPA of PF4V1 increased from 0.002%in pure plasma to 2.356% in NaY-PPC,ACTB increased from 0.003% to 1.633%,the relative abundance order of PF4V1 improved from 312nd in pure plasma to 6th in NaY-PPC and ACTB improved from 294th to 9th(Table S2).Moreover,many of these proteins are closely linked to the development of disease.These phenomena are consistent with previous findings[41,42],that is,the abundance profile of proteins in serum or plasma does not directly affect the composition of proteins in the corona.This occurrence may be due to the fact that corona formation is a complex dynamic process,where lots of different proteins are competing for a limited available surface area[43,44].Higher concentrations of proteins may bind to the particles first,but over time,they will be replaced by proteins with a higher affinity for the surface[45].Moreover,the rich surface properties of NaY give it the ability to adsorb thousands of proteins by utilizing multiple forces [26-29],which facilitates the expansion of the detection of the plasma proteome.In other words,the quantitative composition of NaY-PPC is not only related to the physicochemical properties of NaY,but also closely correlated with the abundance of proteins with affinity on the surface of NaY.
To better understand the qualitative and quantitative performance of our method for proteins of different abundance,the 100 most abundant proteins in the plasma control were divided into different intervals based on their abundance.As shown in Fig.5C,the proportions of these proteins in the plasma control and the NaY-PPC were compared,respectively (Supplementary data 4).The 20 proteins with the highest abundance in plasma occupied the vast majority of the plasma control,especially the top 10,accounting for 70.98%of all proteins.In contrast,proteins other than these 100 more abundant proteins(“Others”in Fig.5C),that is,the middle- and low-abundance proteins,accounted for only 2.54%.With the adsorption of NaY,the percentage of these 20 proteins decreased significantly,from 83.63% to 25.77%.It is particularly worth highlighting that the percentage of “Others” increased 20-fold (from 2.54% to 54.41%),which is certainly an encouraging sign for a comprehensive exploration of the plasma proteome.Then,the proteins identified from the plasma control and NaY-PPC were sorted by RPA from highest to lowest,respectively,and the cumulative RPA was calculated (Fig.S3;Supplementary data 4).It was shown that the curve slope of NaY-PPC was smaller than that of the plasma control,and the cumulative RPA of 95% corresponded to the 65th protein of the plasma control and the 898th protein of NaY-PPC,respectively.All of the above data indicate that our approach based on NaY-PPC can remove high-abundance proteins to some extent,thus enhancing the detection of lowabundance proteins.
3.5.Sensitivity of identification of plasma proteins
To determine the dynamic range,as well as the detection depth,covered by our method,the identified proteins were mapped against the Human Plasma PeptideAtlas[46](Fig.6;Supplementary data 5).As shown in Fig.6A,this method based on NaY-PPC could quantify most of these proteins and detect many uncatalogued proteins.The relationship between the DIA library and the database(Human Plasma PeptideAtlas)showed a similar pattern,but the DIA library covered more proteins(3029 proteins)(Fig.S4).The Human Plasma PeptideAtlas provides the latest and most comprehensive compilation of proteins based on reports from a large number of published studies on human plasma samples,but these proteins are usually based on traditional assays,such as those that deplete highabundance proteins and use fractionation,so many plasma proteins have not been captured in this database [46,47].The broad and deep coverage provided by NaY-PPC allows for the detection of additional uncatalogued proteins that are not detectable by older detection technologies and may be important in biomarker development.
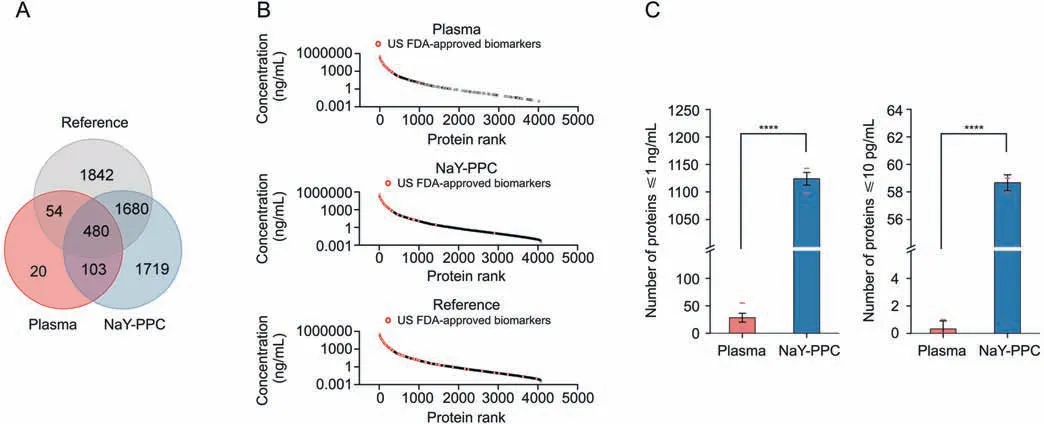
Fig.6.The sensitivity and dynamic range of the identified plasma proteins.(A) Venn diagram of unique and common proteins between NaY-PPC,pure plasma control,and the referenced database (Human Plasma PeptideAtlas).(B) Concentration distribution of plasma proteins.Concentration rankings of proteins from the referenced database (Human Plasma PeptideAtlas)are shown in the bottom panel,and proteins from NaY-PPC and pure plasma are shown in the middle and top panels,respectively.(C)The number of proteins≤1 ng/mL and 10 pg/mL identified from NaY-PPC and pure plasma control.The lower red dash indicates the number of proteins detected in all three replicates,and the upper red dash indicates the number of proteins detected in any of the three biological replicates.Plasma: pure plasma control; NaY-PPC: plasma protein corona formed on zeolite NaY; US FDA: US Food and Drug Administration.****P <0.0001.
In general,the NaY-PPC matching the database extended to almost the entire dynamic range of the database and was mainly concentrated in the low concentration range (≤1 ng/mL).In contrast,proteins from the pure plasma control are mainly those from the high concentration region(>1 ng/mL)with a large number of undetected information gaps in the medium and low concentration regions (Fig.6B).There was an 8-order of magnitude difference between the highest concentration protein(Ceruloplasmin,440,000 ng/mL) and the lowest concentration protein (Ring finger protein 213,0.0035 ng/mL)detected from NaY-PPC.Thus,the NaYPPC-based plasma proteomics analysis approach is able to capture plasma protein information across multiple orders of magnitude without complex workflows.Further,statistics revealed that 1143 proteins ≤1 ng/mL and 59 proteins ≤10 pg/mL could be identified from NaY-PPC,whereas the control group could only capture 55 and 1 protein,respectively (Fig.6C).It should be emphasized that the reproducibility and stability of NaY-PPC remained excellent,even for proteins at concentrations ≤10 pg/mL,whereas the pure plasma control was unreliable for detecting proteins ≤1 ng/mL,let alone 10 pg/mL,which could not even be detected(Fig.S5).The detection and quantification of protein biomarkers is a key application of indepth proteomic analysis.We matched the identified proteins to known biomarkers (109 proteins) approved by the US Food and Drug Administration (FDA) [48].There were 52 US FDA-approved biomarkers identified from NaY-PPC(Fig.6B).
Overall,the above findings suggest that NaY-PPC can minimize the “noise” from high-abundance proteins while significantly improving the coverage of plasma low-abundance proteins.It is worth noting that the interactions between proteins and a wide variety of particle surfaces are quite complex.Proteins can be enriched or weakly adsorbed by particles,which depends on the abundance and diffusion rate of the protein,as well as the affinity between the protein and the particle.High-abundance proteins may bind to the particle first,but will be replaced over time by proteins with a higher affinity for the surface.As mentioned above,the significant decrease(not depletion)in the relative abundance of albumin after NaY adsorption could be attributed to its weaker affinity for zeolite.However,the interactions between zeolites and different proteins and the magnitude of affinity deserve further indepth investigation later with the help of other means.Next,plasma samples of healthy and cancer patients were used to further evaluate the potential of NaY-PPC to differentiate cancer groups from healthy individuals and to discover pharmacodynamic biomarkers.
3.6.Plasma proteomics analysis of LUAD subjects and healthy individuals
Lung cancer is the highest incidence malignancy worldwide[49],and LUAD is a common subtype of non-small cell lung cancer[50].A comprehensive and precise analysis of the plasma proteomic information obtained from LUAD patients can effectively facilitate the discovery of disease molecular markers and drug targets.Plasma samples from 30 LUAD patients and 15 healthy subjects were detected using a NaY-PPC-based plasma proteomics analysis method.
In total,4804 proteins were identified in 45 discovery cohorts,and 3415 were present in more than 70% of the samples.(Fig.7A;Supplementary data 6).The 3415 proteins identified in 70% of the plasma samples were used for further quantitative analysis.Principal component analysis (PCA) of all plasma samples revealed a clear boundary between tumor patients and healthy individuals(Fig.S6),indicating the development of an abnormal proteomic landscape associated with LUAD progression.There were 470 proteins differentially expressed in the tumor group compared to the health group (P-adjust <0.05,FC (T/H) >2 or <0.5),including 283 proteins upregulated and 187 downregulated in tumors (Fig.7B;Supplementary data 6).PCA analysis based on these proteins could better separate tumor groups from healthy controls(Fig.7C).Many of the differential proteins have been shown to be closely associated with the development of LUAD,such as FCN3[51],KRAS[52],PKM[53],CASP1[54],and PGAM1[55].Specifically,Jang et al.[51]showed that FCN3 was significantly downregulated in cancer tissues,which is in line with our plasma proteomics data(FC=0.163,P-adjust = 0.025),suggesting that FCN3 may be a promising diagnostic and prognostic biomarker.KRAS oncoprotein is a key driver in 33% of LUAD [52].Also,the level of CASP1 expression was strongly correlated with the overall survival rate of LUAD[54].More importantly,some differential proteins not reported to be directly related to LUAD,such as AIP,PPP1R12C,PARK7,HSPB1,and TYMP,are closely connected with the development of other diseases,and the study of these proteins may contribute to our understanding of LUAD.In addition,114 differentially expressed proteins met the condition of receiver operating characteristic-area under the curve(ROC-AUC) ≥0.9.Unsupervised clustering analysis was performed for 45 plasma samples based on the quantitative information of these 114 proteins.As expected,the LUAD and healthy groups could be well separated,indicating the potential of plasma proteomics based on NaY-PPC for biomarker screening in LUAD patients(Fig.7D).
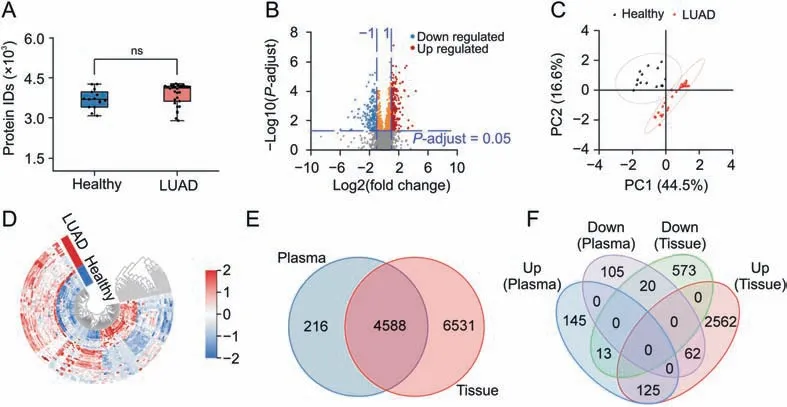
Fig.7.Plasma proteomics analysis of 30 LUAD subjects and 15 healthy individuals.(A)Boxplots of the number of proteins identified in the plasma of healthy individuals and LUAD patients.(B) Volcano mapping of proteins identified in the plasma of healthy individuals and LUAD patients.(C).Principal component analysis (PCA) mapping of the 45 plasma samples based on differentially expressed proteins.(D)Unsupervised clustering of the 45 plasma samples based on 114 differentially expressed proteins with a receiver operating characteristic-area under the curve(ROC-AUC)≥0.9.(E)Venn diagrams of proteins identified in plasma cohorts and tissues.(F)Venn diagrams of differentially expressed proteins in plasma cohorts and tissues.Plasma cohorts in(E,F)refer to plasma samples from 30 LUAD subjects and 15 healthy individuals,and tissues refer to 103 LUAD tumors and their paired non-cancerous adjacent tissues as reported in the work of Xu et al.[56].LUAD: lung adenocarcinoma; Healthy:healthy subjects; Up:up-regulated differential proteins; Down:down-regulated differential proteins.ns: no significant difference.
The findings of our plasma-based proteomics were compared with a recently published tissue-based study of 103 Chinese LUAD patients[56].In Xu et al.'s work[56],11,119 proteins were identified in 103 LUAD tumors and their paired noncancerous adjacent tissues(NAT),including 2,749 upregulated and 606 downregulated proteins.These two independent studies co-identified 4,588 proteins and 220 differentially expressed proteins(Figs.7E and F),indicating that a large number of proteins at the tissue level can be successfully identified in plasma by NaY-PPC,which further demonstrates the reliability and advantages of the NaY-based plasma proteomics approach.More importantly,differentially expressed proteins during disease progression can be identified and detected at the plasma level,which is certainly an encouraging result for early diagnosis and therapeutic detection of diseases.
Overall,the above experimental results demonstrate that changes in plasma proteins caused by the disease may be manifested in protein corona,and NaY-PPC has the potential to successfully reveal subtle changes in health and disease states.The plasma proteomics profiling strategy based on NaY-PPC,as a promising non-invasive technique,will provide a new and advantageous tool for pharmacodynamic biomarkers development,as well as specialized proteomics studies.
4.Conclusions
In summary,a rapid,simple,and reproducible method based on NaY-PPC was first presented for comprehensive and deep plasma protein detection and profiling.This method took only about 6 h to go from proteins to peptides and showed excellent reproducibility with a median CV of 11.58%.NaY could largely eliminate the“masking” effect caused by high-abundance plasma proteins and enhance the detection of low-abundance proteins.The relative abundance of the 20 most abundant proteins in plasma decreased from 83.63%to 25.77%,while that of the middle-and low-abundance proteins increased from 2.54% to 54.41%.With the workflow described herein,approximately 4,000 proteins and 31,000 peptides could be stably quantified in a single plasma sample,covering a dynamic range of 8 orders of magnitude with high sensitivity down to pg/mL.Subtle changes of plasma proteins between healthy individuals and LUAD patients were able to be identified,demonstrating the potential of this methodology to be used in pharmacodynamic biomarker discovery.This method based on NaYPPC achieves an excellent performance of plasma proteome profiling in both depth and breadth,presenting a technological springboard for plasma proteomics exploration and its translational applications.
CRediT author statement
Congcong Ma:Methodology,Validation,Formal analysis,Investigation,Writing - Original draft preparation,Reviewing and Editing,Visualization;Yanwei Li:Resources,Project administration;Jie Li:Resources,Writing - Original draft preparation,Supervision;Lei Song:Software,Data curation,Visualization,Project administration;Liangyu Chen:Software,Data curation,Visualization,Project administration;Na Zhao:Validation,Formal analysis,Visualization,Writing - Reviewing and Editing;Xueping Li:Validation,Formal analysis;Ning Chen:Validation,Supervision;Lixia Long:Validation,Resources;Jin Zhao:Conceptualization,Validation,Investigation,Supervision,Funding acquisition,Writing -Reviewing and Editing;Xin Hou:Validation,Investigation,Supervision,Funding acquisition,Writing - Reviewing and Editing;Li Ren:Conceptualization,Resources,Supervision;Xubo Yuan:Conceptualization,Investigation,Resources,Project administration,Funding acquisition,Writing- Reviewing and Editing.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No:51773151).
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2023.04.002.
 Journal of Pharmaceutical Analysis2023年5期
Journal of Pharmaceutical Analysis2023年5期
- Journal of Pharmaceutical Analysis的其它文章
- DNA barcoding in herbal medicine: Retrospective and prospective
- Benzodiazepines in complex biological matrices: Recent updates on pretreatment and detection methods
- Ginsenoside Rk3 is a novel PI3K/AKT-targeting therapeutics agent that regulates autophagy and apoptosis in hepatocellular carcinoma
- Spatiotemporal pharmacometabolomics based on ambient mass spectrometry imaging to evaluate the metabolism and hepatotoxicity of amiodarone in HepG2 spheroids
- Host cell protein quantification workflow using optimized standards combined with data-independent acquisition mass spectrometry
- A robust luminescent assay for screening alkyladenine DNA glycosylase inhibitors to overcome DNA repair and temozolomide drug resistance