
过表达Miro1 慢病毒载体的构建及Miro1 高表达间充质干细胞稳定转染细胞株的建立与鉴定
2023-06-14 09:39:32陈镜伊赵玲萍刘雯丽习佳飞梁志欣
解放军医学院学报 2023年4期
陈镜伊,赵玲萍,刘雯丽,习佳飞,梁志欣
1 解放军总医院第一医学中心呼吸与危重症医学科,北京 100853;2 军事科学院军事医学研究院辐射医学研究所,北京 100850;3 华南生物医药研究院,华南干细胞与再生医学研究中心,广东广州 510005
Miro1 (mitochondrial Rho GTPase 1,Rhot1)是线粒体转移的关键蛋白之一,是首个被证明可以加速线粒体转移的蛋白。在线粒体的细胞接触依赖性转移中,Miro1 作为一种钙敏感衔接蛋白,可在Miro2、TRAK1、TRAK2、Myo19 等辅助蛋白的协助下将线粒体连接到KIF5 运动蛋白上,从而实现线粒体沿微管在细胞间移动[1-3]。间充质干细胞(mesenchymal stem cell,MSC)具有归巢分化、免疫调节和促进线粒体转移等功能[4-5],其极具潜力的损伤修复及抗炎功能使之成为肺、心脏、肝等多种组织器官损伤的新型治疗策略,是再生医学的重要组成部分。一些研究发现Miro1 蛋白可以促进线粒体转移,从而修复气道上皮细胞、巨噬细胞的线粒体功能障碍,是间充质干细胞修复肺损伤的重要环节之一[6-7]。因此,增强MSC 的线粒体转移能力可能成为优化MSC 治疗效果的潜在途径。
慢病毒载体源于人类免疫缺陷病毒1 型病毒,凭借其病毒持久性独特的复制周期,使宿主细胞稳定表达外源基因[8],目前已广泛应用于基因编辑。我们先前的研究发现慢病毒载体感染MSC 效率较高,是理想的MSC 基因编辑载体。因此,本研究拟采用慢病毒载体构建Miro1 过表达间充质干细胞的细胞株,对Miro1 的表达从mRNA 和蛋白水平进行鉴定,并对转染后的细胞株进行干细胞特性的验证,所得细胞株将从线粒体动力学角度,为研究间充质干细胞的抗炎修复机制提供细胞模型。
材料与方法
1 细胞、试剂与仪器 研究对象为小鼠骨髓间充质干细胞(MUBMX-01001,Oricell)。仪器包括:PCR 仪(C1000,Bio-Rad)、凝胶成像系统(Bio-Rad Gel Doc XR ,Bio-Rad)、低温超速离心机(5819R,Eppendorf)、化学发光成像仪(GE A1680,Amersham Imager 680)、多功能微孔板读板机(DE02223, Molecular Devices® MS)、 CO2孵箱(3111,Thermo Fisher)、荧光倒置显微镜(Nikon,Nikon Ci-S)、小型垂直电泳仪(Bio-Rad)等。试剂包括:质粒小提试剂盒(DP103-03,TiANGEN)、Taq DNA Polymerase (10342020,Fermentas)、dNTP Mix (R1121,Fermentas)、GeneRµLerTM100 bp DNA Ladder (SM0241,Fermentas)、GeneRulerTM1 kb DNA Ladder (SM0311,ThermoFisher)、无内毒素质粒大提试剂盒(DO120,TiANGEN)、Lenti-X p24 Rapid Titer Kit (632200,Clontech)、SYBR® qPCR Mix(THUNDERBIRDTM)、DEPC 水(Solarbio)、电泳缓冲液(Solarbio)、TBST (CWbio)、骨髓间充质干细胞成骨诱导分化培养基试剂盒(MUXMX-90021,Oricell)、骨髓间充质干细胞成脂诱导分化培养基试剂盒(MUXMX-90031Oricell)、Anti-HA tag 抗体(ab9110,Abcam)、GAPDH 一抗、辣根过氧化物酶标记兔抗鼠IgG、蛋白电泳Marker (Thermo Fisher Scientific), 2 × Laemmli Sample Buffer(Bio-Rad)、RIPA 裂解缓冲液(碧云天)、PVDF 膜(Millpore)、10% SDS-PAGE 凝胶超快速配制试剂盒(P0690,碧云天)、限制性内切酶(NEB)、1 ×PBS (Sigma),0.25%胰酶-EDTA 消化液(Sigma)、DMEM 培养基(Sigma)、胎牛血清(Gibco)等。
2 目的基因的选择与PCR 扩增 Miro1 蛋白为RHOT1 基因表达产物。从NCBI 官网获取RHOT1基因mRNA 序列为Rhot1 ras homolog family member T1 [Mus musculus (house mouse)] (NM_021536.8)。根据Gibson 反应原理,设计引物序列见表1。

表1 mRhot1 引物序列Tab. 1 Primer sequences of mRhot1
PCR 反应结束后,对反应产物进行琼脂糖凝胶电泳,并对目的片段切胶回收,测定浓度之后将DNA 放-20℃保存备用。
3 质粒构建与酶切鉴定 按照质粒小提试剂盒说明书进行骨架质粒提取,将PCR 扩增后的基因片段和线性化骨架载体DNA 加入Gibson 反应体系,取适量反应产物转化大肠埃希菌感受态细胞。随机挑取单菌落进行菌落PCR,挑选出能扩增出目标长度的克隆,接种到LB 培养基培养过夜,随后抽提小提质粒DNA 送测序,比对后使用测序正确的初级载体通过LR 反应将目的序列重组到最终骨架载体上从而获得含有目的序列的终载体。对于终载体,采用SnapGene 软件确定酶切方案,使用限制性内切酶对最终载体的质粒DNA 进行酶切,对酶切产物进行琼脂糖凝胶电泳,参照DNA Ladder,能且仅能够切出特定长度DNA 条带的载体为正确的最终载体。
4 构建慢病毒载体 慢病毒载体划线接种,取单菌落接种按照试剂盒说明书进行提取质粒DNA,转染前培养HEK293T 细胞至细胞融合度为80% ~90%,根据脂质体转染法原理配制转染复合物并转染HEK293T 细胞,收集培养上清进行离心、浓缩与纯化。采用ELISA 法对所获得的慢病毒载体进行病毒滴度测定,并用病毒转导HEK293T 细胞根据荧光图初步评估慢病毒载体转导能力。
5 慢病毒感染BMSCs 及细胞株的纯化 感染前1 d 将细胞接种6 孔培养板,感染时细胞的融合率约为50%,感染前需换液,加入1 mL 培养基,病毒冰浴融化后接种相应体积的病毒液及聚凝胺(Polybrene),混匀后放入37℃孵箱中继续培养;4 h 后补充1 mL 培养基,14 h 后换液,此后按常规每3 d 换液;病毒感染72 h 后用倒置显微镜观察荧光,监测感染效率,出现较多荧光时于等量接种的转染BMSC 和未转染BMSC 分别加入等浓度嘌呤霉素,待未转染BMSC 全部死亡并且可观察到满意荧光量时撤去嘌呤霉素,完成纯化。由此可得三组细胞株。(1) BMSC 组:正常骨髓间充质干细胞;(2) Con-BMSC 组:空载病毒载体感染的骨髓间充质干细胞;(3) MiroHi-BMSC组:过表达Miro1 的病毒载体感染的骨髓间充质干细胞。
6 RT-qPCR 检测目的基因的mRNA 表达水平取适量三组细胞进行培养,密度达90%时提取RNA,倒去培养液,PBS 清洗后加入适量TRIzol试剂并充分吹打,转移至Eppendorf 管中,室温裂解5 ~ 10 min。按照比例加入氯仿,剧烈摇动,4℃静置15 min 可见分层后4℃下120 00g 离心15 min。取上清液加入异丙醇,颠倒混匀4℃静置15 min 后,4℃下12 000g 离心15 min。小心吸去上清,用75%乙醇充分重悬洗涤RNA 沉淀。再次离心后弃上清,室温干燥沉淀直至变为透明,用适量DEPC 水溶解RNA。紫外分光光度计测其光密度值,OD260/OD280为1.8 ~ 2.0 者为较为纯净RNA。对所提取的RNA 进行反转录,-20℃保存备用。分别设计RHOT1 和GAPDH 引物:
RHOT1 -sense: 5’-CCTGTGTGGAGTGTTCA GCA-3’
RHOT1-antisense: 5’-TCATTCAGAGTGCCGT CGTT-3’
GAPDH-sense: 5 ’-CCTGTGTGGAGTGTTCA GCA-3’
GAPDH-antisense: 5’-TCATTCAGAGTGCCGT CGTT-3’
合成引物后,将2×SYBR Green PCR Master-Mix、模板DNA、引物及去离子水融化后振荡混匀,根据SYBR@Green 法计算并配制RT-qPCR 反应体系。反应体系见表2。

表2 反应体系构成Tab. 2 Composition of the reaction system
设置PCR 仪程序,上机进行RT-qPCR 反应。反应条件如下:95℃预变性5 min;95℃变性30 s,54℃退火30 s,72℃延伸30 s,共35 个循环;72℃延伸10 min,12℃回复到室温。反应结束后使用Bio-Rad CFX Manager 及GraphPad Prism 9 进行数据处理分析。
7 Western blot 检测目的基因的蛋白表达水平取适量三组细胞进行培养,密度达90%时提进行蛋白提取,用预冷的PBS 洗涤,加入适量细胞裂解液冰上充分裂解后用细胞刮子分别转移到Eppendorf 管中,4℃下12 000g 离心20min,取上清。按照BCATMProtein Assay Kit 说明书绘制标准曲线,将A 液和B 液按照50∶1 混合,按比例配制混合液与待测蛋白,37℃孵育20 min 后,用595 nm 波长测定吸光度,对蛋白进行定量,根据浓度配平,-80℃保存备用。配制分离胶及浓缩胶后每条泳道10 µL 点样,配制电泳液组装电泳装置进行电泳,待条带分离适当后停止,“三明治法”组装转膜装置,在200 mA、100 min 条件下将条带转至PDVF 膜上,转膜完成后TBST 溶解的5%脱脂牛奶室温封闭1 h,取出PVDF 膜洗净4℃孵育一抗过夜,次日洗膜后将二抗溶于5%脱脂牛奶室温孵育1 h,漂洗后配制显影液ECL 法进行显影。
8 病毒感染后细胞株水平及垂直迁移能力鉴定分别消化、收集BMSC、Con-BMSC、MiroHi-BMSC三种细胞,调整密度为5×105/孔加入6 孔板中,细胞长满后进行划痕实验检测RHOT1 基因干扰后的MSC 的水平迁移能力:用10 µL 的枪头在培养孔中央划痕,用PBS 轻轻洗涤以清除被划下的细胞,无血清孵育10 h,取出显微镜下观察并拍照记录,比较划痕宽度的变化。以5×104/孔的密度接种在24 孔板中的Transwell 小室上室,在小室下的培养板中加入等量MSC 完全培养基,以检测RHOT1 基因干扰后的MSC 的垂直迁移能力。12 h后,对细胞进行固定和结晶紫溶液染色,清除多余染料后观察、拍照。
9 病毒感染后细胞株成骨及成脂诱导分化能力鉴定 取三种BMSC 分为相同的两组别进行诱导分化,待细胞达到80% ~ 90%融合时,一组进行成骨诱导分化:加入2 mL/孔的成骨诱导分化完全培养基,放入37℃、5% CO2培养箱中培养,每3 d换液,镜下可观察到明显钙结节时,吸出培养基,用PBS 轻缓漂洗3 次,用4%多聚甲醛溶液固定30 min,PBS 漂洗3 次,每孔加入2 mL 茜素红工作液染色15 min,PBS 漂洗3 次后显微镜观察、拍照;另一组进行成脂诱导分化:加入2 mL/孔的成脂诱导完全培养基A 开始诱导,2 ~ 3 s 后将培养基更换为维持培养基B,维持24 h 后,再次换为成脂诱导完全培养基A,如此进行循环;镜下可观察到明显脂滴时,小心轻缓倒去培养夜,加入PBS 轻缓漂洗3 次,4%多聚甲醛固定30 min;然后用PBS 轻缓漂洗3 次,预热过滤处理油红O 工作液,2 mL/孔染色15 min;用PBS 漂洗3 次后显微镜观察、拍照。
10 统计学分析 实验重复3 次,采用SPSS 18.0软件及GraphPad Prism 9 进行数据分析及整理。计量资料以±表示,两组比较采用t检验,多组比较采用单因素方差分析,然后采用Dunnett-t进行事后多重比较。P<0.05 为差异有统计学意义。
结 果
1 载体设计图谱及质粒构建鉴定 过表达载体选取EF1A 和CMV 启动子分别启动mRhot1 和EGFP,在目的序列加入HA 标签蛋白序列,对照空载体采用双荧光,即EF1A 和CMV 启动子分别启动mCherry 和EGFP。由此,过表达载体为pLVEGFP:T2A:Puro-EF1A>mRhot1[NM_021536.8]/HA,空载体为pLV-EGFP:T2A:Puro-EF1A>mCherry,载体图谱如图1 所示。本研究采用Gateway 技术构建载体,经酶切鉴定结果如图2 所示,所构建质粒经酶切后可产生3 305 bp、3 985 bp、1 246 bp、2 761 bp 的特异性条带,提示我们构建的质粒成功插入了mRhot1[NM_021536.8]片段。

图1 过表达载体与对照空载体设计图谱A:pLV-EGFP:T2A:Puro-EF1A>mRhot1[NM_021536.8]/HA;B:pLV-EGFP:T2A:Puro-EF1A>mCherryFig.1 Gene mapping of the overexpression and control vectorsA: pLV-EGFP:T2A:Puro-EF1A>mRhot1[NM_021536.8]/HA; B: pLV-EGFP:T2A:Puro-EF1A>mCherry
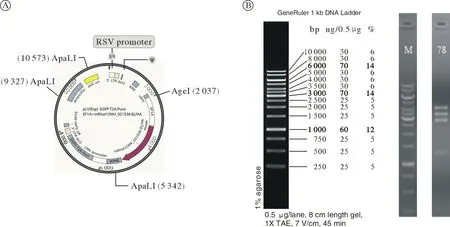
图2 终载体的酶切鉴定结果A:SnapGene 软件查找酶切位点;B:限制性内切酶酶切产物琼脂糖凝胶电泳Fig.2 Identification of the final vector by enzyme digestionA: Finding the digestion sites by the SnapGene software; B: Agarose gel electrophoresis map of the products from the enzyme digestion
2 慢病毒载体构建后感染HEK293T 细胞 对所获得的慢病毒载体进行病毒滴度测定,ELISA 测得病毒滴度为1.45×108TU/mL (Transduction Units per mL,指可以转导并进入细胞中的病毒基因组数量,为慢病毒的滴度测定的常用单位)。随后以MOI=5 感染HEK293T 细胞,感染48 h 后荧光倒置显微镜下可见转染效率达90%以上(图3),故慢病毒载体构建成功且具有较为理想的感染能力。
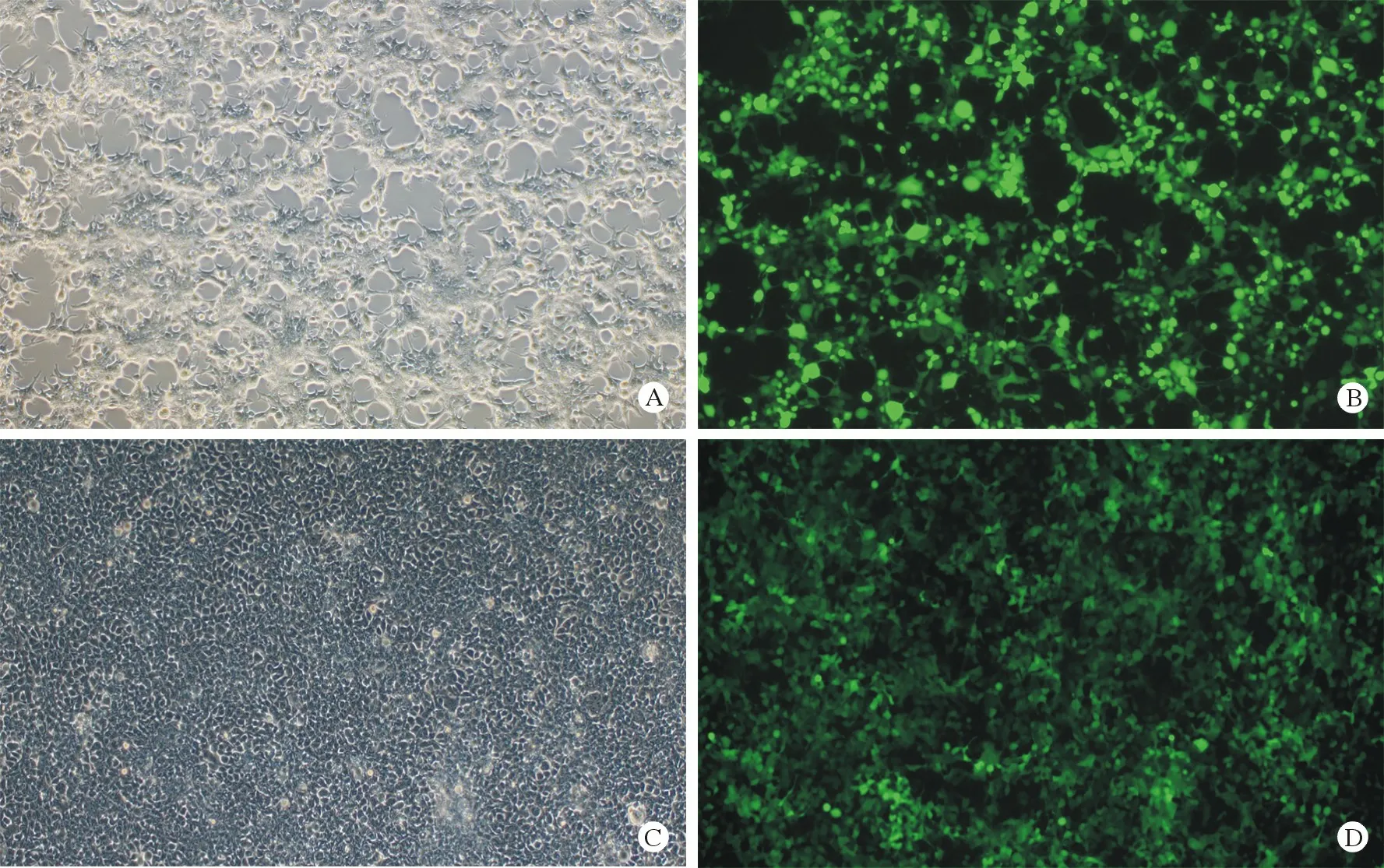
图3 病毒载体感染HEK293T 细胞后48 h 荧光图(100×)A:质粒DNA 转染HEK293T 细胞包装病毒明场图;B:质粒DNA 转染HEK293T 细胞包装病毒EGFP 荧光图;C:病毒感染HEK293T 细胞明场图;D:病毒感染HEK293T 细胞EGFP 荧光图(MOI=5)Fig.3 Photos of HEK293T cells taken by fluorescence microscopy after 48 h of transduction (100×)A: Plasmid DNA transfected virus packaged by HEK293T cells which observed through bright-field microscope; B: Plasmid DNA transfected virus packaged by HEK293T cells which observed through fluorescence microscopy; C: The virus transduced HEK293T cells which observed through bright - field microscope; D: The virus transduced HEK293T cells which observed through fluorescence microscopy (MOI=5)
3 构建Miro1Hi-BMSC 及Con-BMSC 细胞株以MOI=40 进行BMSC 感染,每24 h 镜下观察荧光量,72 h 后开始出现少量荧光,出现较多荧光时进行嘌呤霉素药筛,图4A、图4B、图4C分别为对照病毒载体感染BMSC (Con-BMSC)的明场图、mCherry 荧光图、EGFP 荧光图,图4D、图4E 分别为Miro1 过表达病毒载体感染BMSC(MiroHi-BMSC)的明场图和EGFP 荧光图。可见几乎所有细胞均明显表达mCherry,启动子为EF1A;而EGFP 表达较弱,启动子为CMV,这表明EF1A启动子更适用于Miro1 过表达细胞系的构建。

图4 慢病毒载体转染BMSCs 荧光图(MOI=40,转染11 d,药筛6 d,200×)A:对照慢病毒载体转染BMSCs 明场图;B:对照慢病毒载体转染BMSCs mCherry 荧光图;C:对照慢病毒载体转染BMSCs EGFP 荧光图;D:Miro1 过表达载体转染BMSCs 明场图;E:Miro1 过表达载体转染BMSCs EGFP 荧光图Fig.4 Photos of BMSCs taken by fluorescence microscopy after transduction of lentiviral vectors (MOI=40, 11 days after the transduction,6 days after screening test by puromycin, 200×)A: Control lentiviral vectors transduced BMSCs which observed through bright-field microscope; B: Control lentiviral vectors transduced BMSCs which observed through mCherry fluorescence microscopy; C: Control lentiviral vectors transduced BMSCs which observed through EGFP fluorescence microscopy; D: Miro1 protein overexpressed lentiviral vectors transduced BMSCs which observed through bright-field microscope; E: Miro1 protein overexpressed lentiviral vectors transduced BMSCs which observed through EGFP fluorescence microscopy
4 BMSC、Con-BMSC 和Miro1Hi-BMSC 细胞株的Miro1 表达水平鉴定 提取三组细胞的总RNA和蛋白后进行RT-qPCR 和Western blot,结果如图5。通过对Miro1 蛋白的mRNA 水平的检测,我们发现Miro1Hi-BMSC 组表达Miro1 蛋白的mRNA 水平显著高于BMSC 组和Con-BMSC 组,为BMSC 组的2.27 倍(P<0.05),而BMSC 组和Con-BMSC 组该mRNA 水平差异无统计学差异(P>0.05)。通过对HA 标签蛋白的检测,我们发现Miro1Hi-BMSC 组HA 标签蛋白呈显著强表达,BMSC 组和Con-BMSC 组均无HA 标签蛋白表达,即Miro1Hi-BMSC 组Miro1 蛋白的表达水平显著高于BMSC 组和Con-BMSC 组。
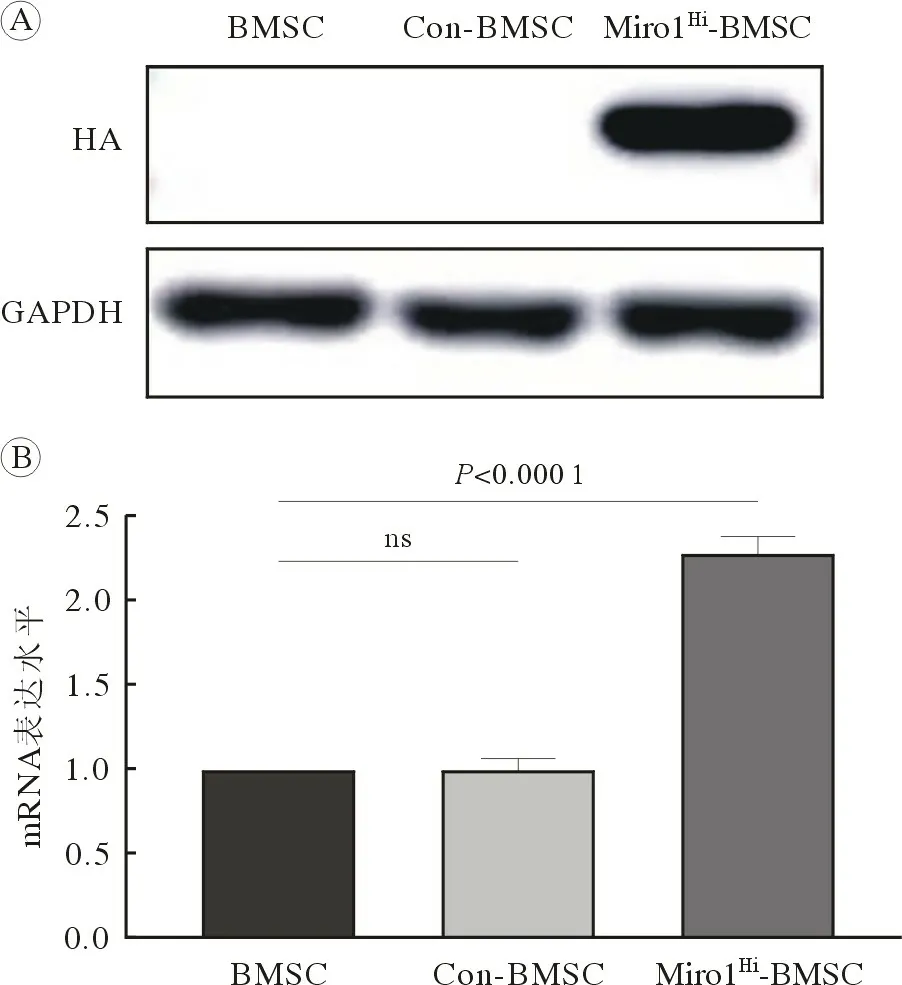
图5 BMSC、Con-BMSC 及Miro1Hi-BMSC 细胞株的Miro1 表达水平鉴定A:蛋白水平Western blot 结果;B:mRNA 水平RT-qPCR 结果Fig.5 Identification of Miro1 expression levels among groups of BMSC, Con-BMSC and Miro1Hi-BMSCA: Western blot results of protein; B: RT-qPCR results of mRNA
5 慢病毒介导的基因干扰对干细胞水平及垂直迁移能力影响的鉴定 通过对BMSC、Con-BMSC和Miro1Hi-BMSC 三组细胞进行水平和垂直迁移实验,结果如图6,三组细胞水平迁移实验10 h 划痕宽度变化量差异无统计学意义(P>0.05),垂直迁移实验24 h 细胞迁移数量差异无统计学意义(P>0.05),这表明慢病毒介导的基因干扰对干细胞水平和垂直迁移能力无显著影响,所构建的Miro1Hi-BMSC 细胞株仍具有与BMSC 基本等同的迁移能力。
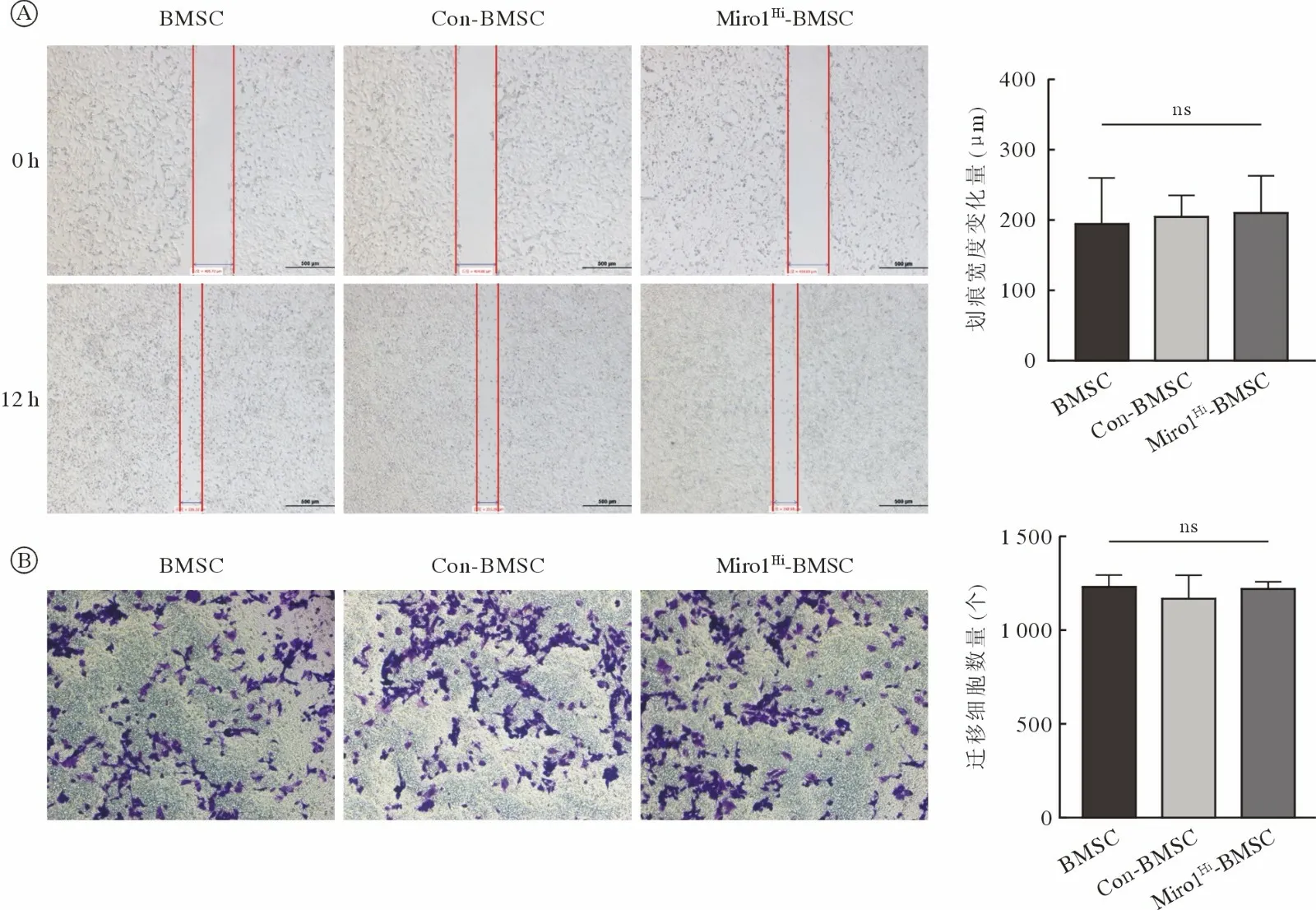
图6 BMSC、Con-BMSC 及Miro1Hi-BMSC 细胞株的迁移能力鉴定A:水平迁移实验结果及统计分析(40×),P>0.05;B:垂直迁移实验结果及统计分析(100×),P>0.05Fig.6 Migration capacity identification among groups of BMSC, Con-BMSC and Miro1Hi-BMSCA: Results of scratch test and statistical analysis (40×), P>0.05; B: Results of vertical migration test and statistical analysis (100×), P>0.05
6 慢病毒介导的基因干扰对干细胞成骨及成脂分化能力影响的鉴定 通过对BMSC、Con-BMSC和Miro1Hi-BMSC 三组细胞进行成骨及成脂诱导分化实验,结果如图7。成骨诱导培养5 d 细胞体积逐渐增大,呈短梭形;诱导10 d 左右,胞质内颗粒显著增多,细胞呈集落样生长,可见散在钙质沉积,细胞结节中心的细胞逐渐融合失去细胞结构,26 d 后经茜素红染色呈红色结节。成脂诱导分化3 d 细胞内可见小脂滴,约15 d 后脂滴数量增加并相互融合,细胞由长梭形变为圆形或多边形,20 d 时进行油红O 染色显示有大量脂质沉淀,BMSC、Con-BMSC、Miro1Hi-BMSC 均可成功诱导成骨、成脂分化。

图7 BMSC、Con-BMSC 及Miro1Hi-BMSC 细胞株的分化能力鉴定A:成骨诱导分化26 d (200×);B:成脂诱导分化20 d (400×)Fig.7 Differentiative capacity identification among groups of BMSC, Con-BMSC and Miro1Hi- BMSCA: 26 days after osteogenesis induces differentiation (200×); B: 20 days after adipogenesis induces differentiation (400×)
讨 论
本研究采用双荧光表达的对照病毒载体对BMSCs 进行感染测试,根据两种不同荧光强度选择出BMSC 中过表达所适用的启动元件,由此确定目的基因的启动子为EF1A。根据设计好的载体图谱,通过经典Gatway、Gibson 法等方法构建Miro1 蛋白过表达的慢病毒载体,并且选取HEK293T 细胞作为转染对象初步测试慢病毒载体感染效率。接下来感染BMSCs 构建Miro1 蛋白过表达的BMSCs 细胞株,通过RT-qPCR 及Western blot 实验发现所构建的Miro1Hi-BMSC 细胞株Miro1表达量显著高于正常BMSC 和Con-BMSC。由于构建细胞株过程中涉及慢病毒转导及基因干扰,我们对所得Miro1Hi-BMSC 和Con-BMSC 细胞株进行干细胞特性检测,通过与正常BMSC 进行对比,我们发现病毒感染后的Miro1Hi-BMSC 和Con-BMSC 细胞株仍具有与正常BMSC 基本相同的迁移能力和分化能力,由此我们成功构建出了保留干细胞特性的Miro1Hi-BMSC 细胞株。
大量研究已验证,传统的慢病毒载体稳定表达转基因方法通常在干细胞中产生稳定的整合。然而,外源基因在干细胞的表达仍可出现降低的现象。研究发现某些沉默元件、组蛋白修饰和DNA 甲基化途径可能干扰了慢病毒载体的表达[9],基因重编程细胞染色质的相关研究表明整合到宿主细胞中的外源基因启动子存在不同程度的表观遗传修饰,影响外源基因转录调控网络[10-11],进一步解释了这种现象的可能机制。因此,设计选择过表达目的基因的启动元件对稳转株的构建具有重要意义。CMV 为常用启动元件,在HEK293T细胞中的高表达已被确定,然而在干细胞却呈现低表达。本研究也发现,与EF1A 启动子相比,CMV启动子表现出低于EF1A 的表达水平。Wen 等[12]发现MSC 中CMV 启动的基因表达减少可能是由DNA 甲基化、组蛋白去乙酰化引起的,胚胎干细胞的相关研究也发现CMV 启动子转基因沉默更显著[13]。 Li 等[14]发现EF1A 启动子代替CMV 启动子可以避免干细胞中CMV 启动子的潜在沉默问题。由此可见,对于构建MSC 稳转株而言,与CMV 相比,EF1A 可能是更为理想的启动元件。
目前,Miro1 相关干细胞基因修饰的已应用于多领域的线粒体动力学研究。Ahmad 等[6]构建过表达Miro1 的骨髓间充质干细胞用于体内和体外实验对线粒体的运输机制展开分子水平的研究,通过免疫荧光可视化线粒体转移转移过程、检测线粒体功能及炎症因子水平,发现Miro1 过表达的间充质干细胞具有更为优秀的气道炎症治疗效果。Tseng 等[15]通过共培养实验发现Miro1 过表达的间充质干细胞可增加受损的神经元的线粒体转移,并在体外氧化损伤后改善神经元代谢,这一过程与TNFAIP2 的表达密切相关。此外,Miro1与线粒体自噬亦具有广阔的研究前景[16],研究发现Miro1 是线粒体自噬的启动蛋白(Parkin)在线粒体上的钙敏感对接位点,不仅存在调控线粒体转移的关键结构域,还参与减轻神经元细胞的线粒体自噬[17-18]。Hsieh 等[19]进一步发现Miro1 的滞留可影响线粒体自噬,从而导致细胞运动缺陷和多巴胺能神经变性,这表明基于Miro1 的深入研究可能为帕金森疾病的治疗开辟新的途径[20]。不仅如此,Miro1 蛋白似乎还参与了线粒体结构的调节,Conejeros 等[21]发现Miro1 蛋白可以调节线粒体融合裂变平衡,参与心肌细胞肥大的发生发展。由此可见,过表达Miro1 细胞株的构建在线粒体自噬和结构领域亦有潜在研究前景。
综上所述,本研究通过慢病毒载体成功建立Miro1 稳定过表达BSMC,该细胞株同样保留有组织迁移、分化修复的干细胞特性,可作为细胞模型研究线粒体的接触依赖性转移过程,且具有潜在的调控线粒体自噬研究前景,推进干细胞治疗的研究进程,为再生医学药物的研发提供参考。
作者贡献陈镜伊:完成实验,数据分析处理,文章撰写;刘雯丽:协助实验,数据分析处理;赵玲萍:技术方法指导;习佳飞:技术方法指导,文章审阅;梁志欣:总体构思,资金资助。
利益冲突所有作者声明无利益冲突。
数据共享声明本篇论文相关数据可依据合理理由从作者处获取,Email:chenjingyi301@163.com。
猜你喜欢
海洋通报(2021年1期)2021-07-23 01:55:14
昆明医科大学学报(2021年3期)2021-07-22 07:39:10
昆明医科大学学报(2021年5期)2021-07-22 07:32:50
现代临床医学(2021年2期)2021-03-29 05:32:44
生物学通报(2021年4期)2021-03-16 05:41:26
世界科学技术-中医药现代化(2021年10期)2021-03-02 05:52:12
医学研究杂志(2015年11期)2015-06-10 06:44:03
中国当代医药(2015年16期)2015-03-01 02:03:11
中国医药导报(2015年27期)2015-02-28 22:08:02
癌变·畸变·突变(2014年2期)2014-03-01 04:39:42
