
葫芦科作物基因组学研究进展
2023-06-07 03:44:26闫晋强晏石娟谢大森刘文睿
广东农业科学 2023年4期
江 彪,闫晋强,晏石娟,谢大森,刘文睿,王 敏
(1.广东省农业科学院蔬菜研究所/广东省蔬菜新技术研究重点实验室,广东 广州 510640;2.广东省农业科学院农业生物基因研究中心,广东 广州 510640)
葫芦科(Cucurbitaceae)是世界上最重要的食用植物科之一,有95 个属900 多个种,其重要性仅次于禾本科、豆科和茄科,位居植物界第四。据联合国粮食及农业组织(Food and Agriculture Organization of the United Nations,FAO)2019 年统计显示,世界葫芦科作物的栽培面积约400 万hm2,年产值约3 000 亿元。葫芦科包含许多重要作物,包括黄瓜(Cucumis sativus)、甜瓜(Cucumis meloL.)、西瓜(Citrullus lanatus)、南瓜(Cucurbitaspp.)、冬瓜(Benincasa hispida)、丝瓜(Luffa aegyptiaca)、苦瓜(Momordica charantiaL.)等常见的蔬菜和瓜果,在农业生产和人民生活中均占有举足轻重的地位[1-2]。葫芦科作物不仅是人们重要的饮食来源,许多还具有独特的食疗保健价值。冬瓜富含丙醇二酸,可抑制人体内糖转化为脂肪,能有效防止体内脂肪堆积,对于肾病、高血压、浮肿患者大有益处[3]。苦瓜富含皂苷,具有明显的降血糖、减肥、抗氧化等多种药理作用[4-7]。
全基因组测序,就是一次性测定一个生物体基因组全部DNA 序列的过程。自2004 年以来,以Illumina 边合成边测序为代表的高通量测序技术逐渐发展成熟,测序通量大幅提升、成本急剧下降,其大规模商业化应用促成数以千计的生物体完成了全基因组测序[8]。近年来,以Pacific Biosciences 单分子实时测序和Nanopore 纳米孔测序为代表的长读长测序技术得到快速发展,为基因组从头组装以及结构变异检测、泛基因组学研究等提供了极大便利[9]。
基因组从头组装(de novo assembly)是指在不需要任何参考序列的情况下,将生物体基因组测序产生的序列进行拼接、组装,从而绘制该生物体的全基因组序列信息。基因组从头组装过程,可简单描述为测序片段(reads)组装形成重叠群(contig)、重叠群连接产生支架片段(scaffold),最后再借助遗传图谱、光学图谱等进行锚定产生染色体(chromosome)。基因组从头组装的核心算法主要可以分为几类:基于贪婪算法(Greedyextention)、基于Overlap-Layout-Consensus(OLC)、基于de Bruijn Graph,以及上述两种或多种算法的组合[10]。在这些算法的基础上,大量研究者开发出多种适用于不同测序平台的组装软件和流程,如适用于Illumina 测序平台的Velvet、SOAPdenovo、ABySS 等[11],以及针对长读长测序或者混合测序数据的Canu、Flye、Hifiasm 等[12]。随着测序技术、组装算法的不断发展,组装质量也不断提升,越来越多的生物体基因组测序实现了染色体水平组装。截至目前,葫芦科作物中的黄瓜、甜瓜、西瓜、南瓜、苦瓜、葫芦、冬瓜、丝瓜、蛇瓜、佛手瓜等均完成了全基因组测序(表1),显著提升了葫芦科作物基因组学、系统进化和分子生物学等领域的研究水平[13-14]。本文简要回顾了葫芦科主要作物的全基因组测序进展,并总结归纳了全基因组测序在葫芦科作物起源与进化关系、重要农艺性状基因挖掘等方面的实际应用,旨在为葫芦科作物基因组学相关研究提供重要的参考依据。
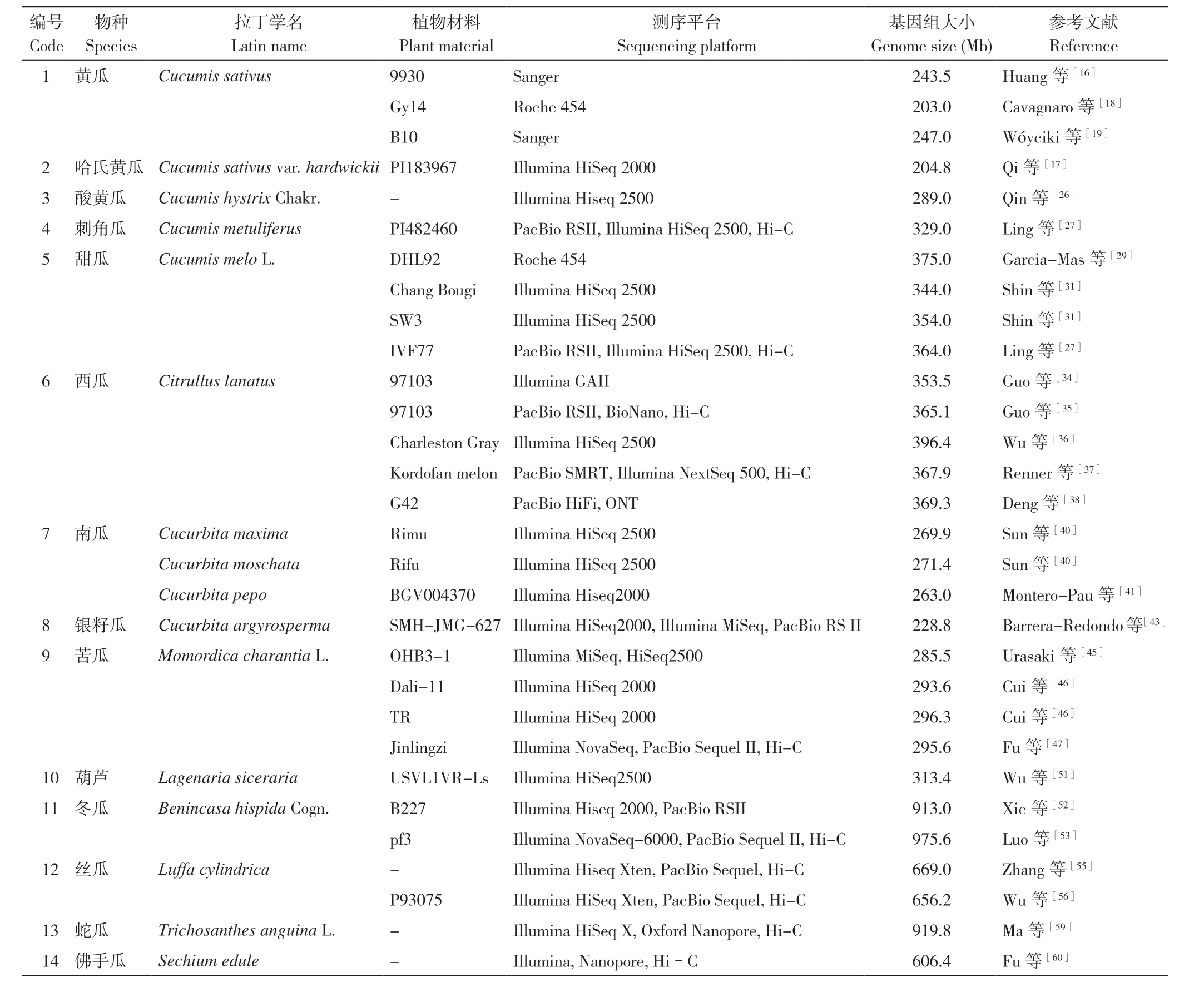
表1 已测序葫芦科作物基因组列表Table 1 List of genome-sequenced Cucurbitaceae crops
1 葫芦科作物基因组测序
1.1 黄瓜基因组测序
1.1.1 栽培黄瓜 黄瓜(Cucumis sativus,2n=2x=14)起源于喜马拉雅南麓的热带雨林地区,是国际上最重要的蔬菜作物之一,也是分子生物学研究的重要模式作物。在葫芦科作物中,黄瓜的染色体数最少,其基因组大小约376 Mb[15]。2007年初,中国农业科学院蔬菜花卉研究所发起国际黄瓜基因组计划,这是我国发起并主导的第一个大型植物基因组计划。该研究利用Sanger 测序方法对华北密刺类型黄瓜高代自交系9930 进行全基因组测序,测序深度为72.2×,组装获得的基因组为243.5 Mb,约占其基因组的66.3%。在黄瓜基因组中共预测26 682 个蛋白编码基因,每个基因的长度平均为1 046 bp,其中20 000 多个基因被定位到染色体上[16]。基于9930 参考基因组,Qi 等[17]从世界范围内3 342 份黄瓜种质资源中筛选出115 份核心种质,并对其进行深度重测序,构建了一张包含360 多万个位点的全基因组遗传变异图谱;在遗传变异图谱中共鉴定到112 个假定的驯化区段,其中1 个区段含有1 个与果实苦味丢失有关的基因,丢失苦味是黄瓜重要的驯化特征。此外,通过研究栽培群体之间的基因组差异,Qi 等[17]在β-胡萝卜素羟化酶基因中发现1 个自然变异,可用于培育营养价值更高的黄瓜品种。
随后,学者们陆续完成不同品系黄瓜的基因组测序。美国威斯康星大学麦迪逊分校以典型北美加工类型黄瓜品系Gy14 为材料,利用Roche 454 平台,采用从头测序策略进行基因组测序,测序深度为36×,组装获得4 219 个scaffolds,其基因组序列总长为203.0 Mb[18]。Wóyciki 等[19]以北欧Borszczagowski 品种B10 为材料,利用Sanger 测序方法进行全基因组测序,组装获得247 Mb 的基因组序列。为提高Gy14 基因组质量,Yang 等[20]将173.1 Mb 的Gy14 基因组和193.3 Mb 的9930 基因组挂载到同一个遗传图谱上,利用9930 基因组的scaffold 填补Gy14 基因组中相邻scaffolds 间的空隙,最终获得192.6 Mb 的Gy14基因组(占基因组的53.0%),其中19.5 Mb 的序列来源于9930 的基因组序列。
1.1.2 哈氏黄瓜 哈氏黄瓜(C.sativusvar.hardwickii,2n=2x=14)是目前公认的栽培黄瓜的野生祖先,具有较好的抗逆和抗病虫能力[21]。Qi等[17]以哈氏黄瓜PI183967(CG0002)为材料,利用Illumina 测序技术进行从头测序,共组装获得204.8 Mb 基因组序列,其scaffold N50 为4.2 Mb,并预测了23 836 个蛋白编码基因。通过与9930基因组进行对比,共鉴定到21 021 个直系同源基因[17]。
1.1.3 酸黄瓜 酸黄瓜(C.hystrixChakr.,2n=2x=24)是黄瓜的野生近缘种,具有耐低温弱光、高抗霜霉病、抗线虫等优良性状[22-25]。酸黄瓜基因组约为416 Mb,大于黄瓜基因组,但小于甜瓜基因组[26]。Qin 等[26]利用Illumina 测序平台完成酸黄瓜全基因组测序,组装获得的基因组约为289 Mb,预测了23 864 个蛋白编码基因。通过全面的比较基因组分析发现,与甜瓜相比,酸黄瓜在系统发育上与黄瓜更为接近,为酸黄瓜与黄瓜成功杂交奠定了分子基础。此外,Qin 等[26]还发现酸黄瓜基因组中富含“防御响应”基因,共有104 个编码抗病基因类似物的核苷酸结合位点。
1.1.4 刺角瓜 刺角瓜(C.metuliferus,2n=2x=24)又名非洲角黄瓜,是黄瓜属野生种,具有较强的抗病虫害能力。刺角瓜基因组约368 Mb,Ling等[27]以CM27(PI482460)为材料,利用PacBio SMART 测序平台进行全基因组从头测序,测序深度为93×,组装获得329 Mb 的基因组序列,其N50 序列长度为2.9 Mb,共预测了29 214 个蛋白编码基因。随后,通过Hi-C 组装和人工校正,共有316.82 Mb 的CM27 基因组序列被挂载到12条染色体上[27]。系统发育分析结果表明,刺角瓜与甜瓜的分化时间在1 780 万年以前,通过刺角瓜与甜瓜基因组之间的比较,发现这两个物种的8 条染色体存在较大的结构变异[27]。
1.2 甜瓜基因组测序
甜瓜(Cucumis meloL .)是第二个完成基因组测序的葫芦科作物,其染色体数目为2n=2x=24,基因组大小约为450 Mb[28]。2012年,西班牙农业基因组研究中心以甜瓜双单倍体DHL92 为材料,采用罗氏454 平台进行全基因组测序,测序深度为13.52×,组装获得1 594 个scaffolds,序列总长为375.0 Mb(v3.5.1)[29]。其scaffold N50 为4.68 Mb,且最长的78 个scaffolds序列总长占组装基因组的90%,表明该基因组的质量较好。该基因组共预测27 427 个蛋白编码基因,每个基因的平均长度为2 776 bp、外显子为5.85 个[24]。随着生物信息学技术的不断发展,Ruggieri 等[30]对DHL92基因组进行重新组装和注释,发布了序列信息更完整、注释信息更全面的参考基因组v3.6.1 版本,新基因组在基因结构完整性、UTR 区域界线等方面均得到明显提升。
2019 年,韩国学者发布了薄皮甜瓜(C.meloL.var.makuwa)Chang Bougi 和SW3 的基因组序列[31]。其中Chang Bougi 是韩国地方甜瓜品种,组装的基因组为344 Mb,scaffold N50 为1.0 Mb,预测了36 235 个蛋白编码基因。SW3 是来源于农友生物公司(NongWoo Bio Company)的高品质材料,其组装的基因组为354 Mb,scaffold N50 达到1.6 Mb,预测了38 173 个蛋白编码基因[31]。此外,Ling 等[27]以北京薄皮甜瓜地方种IVF77 为材料,组装获得染色体水平的参考基因组,其测序深度为84×,组装获得364 Mb 的基因组,其中339.72 Mb 的基因组挂载到12 条染色体上,并预测了27 073 个蛋白编码基因。薄皮甜瓜参考基因组的绘制,为其果实发育、抗病机理及品种选育等研究奠定了基础。
为探明甜瓜的遗传基础和驯化历史,中国农业科学院郑州果树研究所联合国内外单位,构建了世界上第一个甜瓜全基因组变异图谱[32]。该研究分析了1 175 份甜瓜种质资源的基因组变异,共鉴定了560 万个SNP;在此基础上,发现甜瓜可能发生过3 次独立的驯化事件,一次发生在非洲地区,另外两次发生在亚洲地区,并分别产生厚皮甜瓜和薄皮甜瓜两个栽培亚种;同时,通过全基因组关联分析(Genome wide association study,GWAS)等手段,定位了200 余个与甜瓜苦味、酸味、果实大小、果肉颜色等性状相关的候选基因和位点[32]。东北农业大学甜瓜团队进一步研究发现,同为栽培类型的地方品种和改良品种在群体结构和进化方面存在明显差异,提出野生材料-地方品种-改良品种的“Two-step”独立驯化模式,并结合选择压力分析和全基因组关联分析鉴定到8 个影响果实性状的改良位点[33]。
1.3 西瓜基因组测序
西瓜(Citrullus lanatus)是世界性重要水果之一,其染色体数目为2n=2x=22,基因组大小约为425 Mb[28]。Guo 等[34]以东亚西瓜品种97103为材料,利用Illumina 测序技术获得46.18 Gb 的高质量基因组,测序深度达到108.6×,通过从头组装产生353.5 Mb 的基因组(97103v1),占其基因组的83.2%,共预测了23 440 个蛋白编码基因。组装的基因组由1 793 个scaffolds 构成,其scaffold N50 长度为2.38 Mb,其中234 个scaffolds挂载到西瓜11 条染色体上,其序列总长约为330 Mb(占组装基因组的93.5%)。通过对3 个不同西瓜亚种的20 份种质进行重测序,获得多个单倍型,确定了西瓜种质的遗传多样性和种群结构,并鉴定了在驯化过程中有限选择的基因组区域[34]。
Guo 等[35]进一步利用PacBio 测序平台对西瓜品种97103 进行长读长测序,结合BioNano 光学图谱和Hi-C 染色体构象捕获技术,绘制了全新一代西瓜高质量基因组精细图谱(97103v2)。组装的基因组大小为365.1 Mb,scaffold N50 为21.9 Mb,预测了22 596 个蛋白编码基因。其中31 个scaffolds 构成11 条染色体,共362.7 Mb,覆盖西瓜组装基因组的99.3%。同时,Guo 等[35]还对414 份代表西瓜属所有现存物种的材料进行全基因组重测序,通过系统进化分析首次明确西瓜7 个种之间的进化关系,发现野生黏籽西瓜是与现代栽培西瓜亲缘关系最近的种群;该研究同时发现了利用野生西瓜进行抗性改良的基因组痕迹,获得与果实含糖量、瓤色、形状等性状关联的43 个信号位点,并鉴定了关键候选基因[35]。
为补充现有参考基因组(97103),Wu 等[36]以美国西瓜栽培种Charleston Gray 为材料,利用Illumina 测序技术进行全基因组测序,测序深度为228×,组装获得396.4 Mb 基因组序列,约占其基因组的94.6%。进一步对美国国家植物种质资源系统中保存的1 365 份西瓜种质进行测序分型(Genotyping-by-Sequencing,GBS),将其分为栽培西瓜、黏籽西瓜和饲用西瓜3 个物种,并从GBS 数据中获得大约25 000 个高质量SNPs[36]。此外,为深入了解从祖先到驯化西瓜的遗传变化,Renner 等[37]以Kordofan melon 为材料,利用PacBio 测序结合Illumina 测序以及Hi-C 绘图技术进行全基因组测序,测序深度约388.8×,组装的基因组包含86 个contigs,总长度为367.9 Mb,N50 为9.34 Mb,并预测了23 043 个蛋白编码基因。在Kordofan melon 和栽培西瓜97103 之间共检测到15 824 个基因组结构变异(Structure Variantions,SVs),并在超过400 份西瓜种质中定位到这些SVs,揭示了等位基因在进化过程中的频率变化[37]。
然而,以上参考基因组仍然有许多缺口。2022 年,北京大学现代农业研究院等单位以小果型西瓜自交系G42 为材料,利用PacBio HiFi 和ONT 数据,结合多种组装策略,完成了端粒到端粒(Telomere-to-Telomere,T2T)无缺口的高质量基因组图谱[38]。组装的基因组总长度为369.3 Mb,预测了24 205 个蛋白编码基因,解析了全部22 个端粒和11 个着丝粒序列信息,同时填补了97103v2 参考基因组所有的220 个缺口[38]。
1.4 南瓜基因组测序
南瓜(2n=2x=40)原产于墨西哥到中美洲一带,在世界范围内普遍种植,包括中国南瓜(Cucurbita maxima)、印度南瓜(C.moschata)、西葫芦(C.pepo)等栽培种[39]。2017 年,中国国家蔬菜工程技术研究中心和美国博伊斯汤普森研究所(BTI)专家合作完成了中国南瓜(基因组约372.0 Mb)和印度南瓜(基因组约386.8 Mb)的全基因组从头测序,测序深度分别为215×和283×,分别组装获得269.9 Mb 和271.4 Mb 的基因组序列,其scaffold N50 分别为4.0 Mb 和3.7 Mb,蛋白编码基因为32 205 和32 076 个[40]。通过与其他瓜类的基因组序列进行进化比较,发现其他瓜类在形成四倍体后会失去部分祖辈基因、重新回到二倍体状态,而南瓜却仍保留四倍体,比较完整地保存了两种祖辈的基因,因此其染色体对数几乎是其他瓜类的2 倍[40]。随后,Montero-Pau 等[41]利用全基因组鸟枪测序方法完成西葫芦(基因组约为283 Mb)全基因组从头测序,组装获得263 Mb 基因组,其scaffold N50 为1.8 Mb,蛋白编码基因共有27 870 个。尽管西葫芦基因组较小,但是其基因组的形成经历了全基因组复制过程[41]。
银籽瓜(C.argyrosperma)是南瓜属另一种重要作物,其基因组约238 Mb[42]。2019 年,墨西哥学者利用Illumina HiSeq2000、Illumina MiSeq和PacBio RS II 等3 个测序平台完成了银籽瓜的全基因组从头测序,Illumina 和PacBio 的测序深度分别为120×和31×,组装的基因组为228.8 Mb,其蛋白编码基因共有28 298 个[43]。葫芦科作物的蛋白编码基因比较和长链非编码RNA(Long non-coding RNA,lincRNA)分析结果表明,南瓜全基因组复制是通过复制基因的新功能化而加快其基因家族的进化速度[43]。
1.5 苦瓜基因组测序
苦瓜(Momordica charantiaL.,2n=2x=22)是一种重要的蔬菜和药用植物,因果实富含具有特殊苦味的三萜化合物而命名[44]。苦瓜基因组约339 Mb,Urasaki 等[45]以苦瓜自交系OHB3-1为材料,通过Illumina 测序,测序深度为110×,从头组装了285.5 Mb 的苦瓜基因组,其scaffold N50 为1.1 Mb,基因组覆盖率约84%。在组装的苦瓜基因组中,共鉴定到45 859 个蛋白编码基因,其数量远高于黄瓜、甜瓜、西瓜等其他葫芦科作物[16,29,34,45]。
随后,Cui 等[46]完成了对苦瓜栽培种Dali-11(基因组约为300 Mb)和野生材料TR(基因组约为300 Mb)的全基因组从头测序,测序深度分别为251×和185×,从头组装的基因组大小分别是293.6 Mb(scaffold N50 为3.3 Mb)和296.3 Mb(scaffold N50 为0.6 Mb)。预测的蛋白编码基因分别为26 427 个(Dali-11)和28 827 个(TR),其数量与黄瓜、西瓜、甜瓜等葫芦科作物的蛋白编码基因相近,但远少于苦瓜OHB3-1 基因组中的数量[16,29,34,45-46]。进一步对从全球16 个国家收集到的187 份苦瓜种质进行重测序,通过基因组遗传多样性分析,发现以TR为代表的21 份野生苦瓜与166 份栽培苦瓜具有显著的遗传分化,推测大果型栽培苦瓜charantia 来源于小果型栽培苦瓜muricata,而不是来源于野生苦瓜macroloba[46]。
2022 年10 月,Fu 等[47]利用Illumina 测序平台完成苦瓜变种金铃子的基因组测序,测序深度为74.31×,组装获得295.6 Mb 的基因组序列。本次组装的金铃子基因组是一个具有高完整性和准确性的端粒至端粒的高质量基因组,其scaffold N50 达到25.4 Mb,蛋白编码基因为19 895 个。在金铃子苦瓜11 条染色体中,有8 条均无gap 存在,其中6 条同时检测到两段的端粒信号[47]。结合转录组和代谢组分析,进一步揭示金铃子果实色素积累与葫芦素生物合成机制[47]。
1.6 葫芦基因组测序
葫芦(Lagenaria siceraria,2n=2x=22)起源于撒哈拉沙漠以南的非洲地区,是葫芦科重要作物,因具有较好的抗病性和耐冷性而常被用作砧木[48-49]。葫芦的基因组约334 Mb[50],Wu 等[51]以葫芦高代自交系USVL1VR-Ls 为材料,利用Illumina HiSeq2500 测序平台进行全基因组从头测序,测序深度为395×,组装获得313.4 Mb 的基因组序列,其N50 为8.7 Mb,共预测到22 472 个蛋白编码基因。Wu 等[51]基于比较基因组学分析确定了葫芦与其他葫芦科作物直接的线性关系以及谱系特异性基因家族的扩增特点,并通过重建葫芦科最新共同祖先的基因组,揭示葫芦科祖先的核型由12 个原染色体和18 534 个原基因组成,且这12 个原染色体大部分保留在目前的甜瓜基因组中,而其他葫芦科作物的基因组则经历了不同程度的重排事件。
1.7 冬瓜基因组测序
冬瓜(Benincasa hispidaCogn.,2n=2x=24)起源于我国南部和印度,广泛分布于亚洲的热带、亚热带及温带地区,是我国最重要的北运菜和度淡蔬菜之一。冬瓜是目前已知葫芦科中基因组最大的作物,达到1.03 Gb[52]。2019 年,广东省农业科学院蔬菜研究所联合中国农业科学院蔬菜花卉研究所等国内外单位,以黑皮冬瓜自交系B227 为材料,利用Illumina 和单分子实时(Singlemolecular real-time,SMAT)测序技术完成冬瓜全基因组从头测序,测序深度为50×,组装获得913 Mb 的基因组序列,其scaffold N50 为3.4 Mb,最长scaffold 为14.5 Mb,并预测了27 467 个蛋白编码基因[52]。通过6 个葫芦科作物的基因组比较分析,揭示冬瓜基因组代表了最古老的核型,并预测祖先基因组拥有15 条始祖染色体。进一步完成146份核心资源的重测序,将其分成野生种(W)、地方种(L)和栽培种(C)等不同类群,其中栽培种又分黑皮冬瓜(C1)和粉皮冬瓜(C2)亚群。同时,构建了一张包含1 600 万个SNP 的基因组变异图谱,发现冬瓜果实变大经历了从野生种到地方种、再到栽培种的两步进化历程[52]。
为丰富冬瓜参考基因组,Luo 等[53]以粉皮冬瓜自交系pf3 为材料,利用PacBio Sequel II 和Illumina NovaSeq-6000 测序平台完成从头测序,测序深度为230×,组装的基因组大小为975.6 Mb,其scaffold N50 高达70.97 Mb,共预测到37 092 个蛋白编码基因,其中85.05%的基因具有功能注释。
1.8 丝瓜基因组测序
丝瓜(2n=2x=26)起源于印度,是一种重要的蔬菜作物,广泛分布于温带和热带地区[54]。我国丝瓜共有2 种,即普通丝瓜(Luff a cylindrica)和有棱丝瓜(L.acutangula)。2020年,河南农业大学以普通丝瓜(基因组约为737 Mb)为材料,结合单分子实时测序(SMRT)、Illumina 测序和Hi-C 等方法,获得74 Gb 的高质量序列,组装的基因组大小为669 Mb,其contig N50 和scaffold N50 分别为5 Mb 和53 Mb,并预测到31 661 个蛋白编码基因[55]。同年,广东省农业科学院蔬菜研究所以普通丝瓜高代自交系P93075 为材料,利用PacBio、Illumina 和Hi-C 技术进行基因组测序,组装的基因组为656.2 Mb,其scaffold N50 为48.76 Mb,共有25 508 个蛋白编码基因[56]。许多与生物和非生物胁迫相关的基因在丝瓜基因组中进行扩增(丝瓜基因组共有462 个NBS-LRR 基因,远多于其他葫芦科作物),该结果与丝瓜的高抗性相一致[56]。
1.9 蛇瓜基因组测序
蛇瓜(Trichosanthes anguinaL.,20=2x=22)原产于印度,世界各地普遍栽培,是集观赏、食用和药用价值于一身的重要葫芦科作物[15,57-58]。Ma 等[59]采用Illumina 测序平台完成蛇瓜基因组从头测序,测序深度为108.5×,并利用Hi-C 技术组装获得919.8 Mb 的基因组,被挂载到11 条染色体上。蛇瓜基因组共注释到22 874 个蛋白编码基因,而重复序列占整个基因组的80.0%[59]。系统发育分析结果表明丝瓜与蛇瓜的近缘关系最为密切,可能于3 300 万~4 700 万年前由它们的共同祖先分化而来[59]。
1.10 佛手瓜基因组测序
佛手瓜(Sechium edule,2n=2x=28)原产于墨西哥,是一种药食兼用型蔬菜作物。佛手瓜基因组大小为710.23 Mb,Fu 等[60]利用Nanopore三代测序技术完成基因组从头测序,测序深度为151×,进一步利用Hi-C 技术组装获得606.4 Mb 的基因组序列,被挂载到14 条染色体上,其scaffold N50 为46.56 Mb,基因组共含有28 237 个蛋白质编码基因。通过与其他物种基因家族比较,发现佛手瓜与蛇瓜的进化关系最为密切,并可能于2 700 万~4 500 万年前由它们的共同祖先分化而来,同时研究发现佛手瓜在2500(±400)万年间发生过一次全基因组复制事件,是葫芦科内的第三次全基因组复制事件,为佛手瓜的基因进化研究提供了理论依据[60]。
2 全基因组测序在葫芦科作物中的应用
2.1 葫芦科作物的起源与进化
在基因组测序开展之前,葫芦科作物的共同原始祖先具有什么特征,不同物种之间的进化关系如何尚不清楚。随着黄瓜、甜瓜、西瓜、南瓜和葫芦参考基因组的陆续发布,人们推测葫芦科作物的祖先含有12条染色体,与甜瓜最为接近[51]。冬瓜基因组测序后,Xie 等[52]发现冬瓜比甜瓜更为保守,是迄今发现拥有最古老基因组的葫芦科作物,并推断它们起源于一个拥有15 条染色体的祖先基因组,经过染色体多次断裂和融合等事件形成目前丰富多样的葫芦科大家族。Guo 等[13]以葫芦科52 个属136 个物种的转录组与基因组数据为基础,确认了葫芦科作物属以上的亲缘关系(图1),揭示葫芦科最近共同祖先起源于白垩纪晚期,推测基因组加倍事件是促使葫芦科作物起源后快速分化的原因。
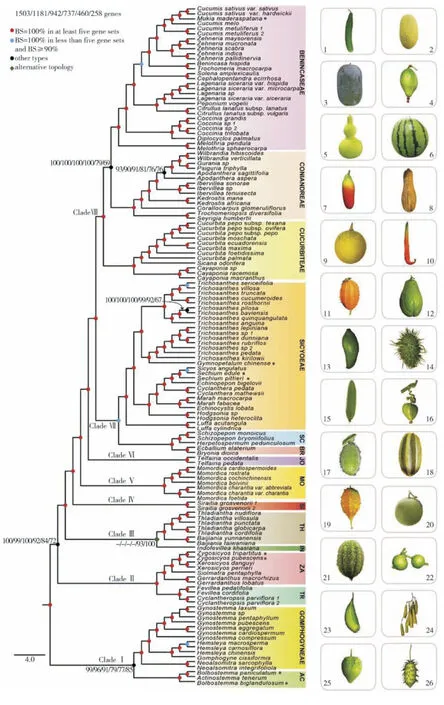
图1 葫芦科作物亲缘关系[13]Fig.1 Relationship between Cucurbitacea crops[13]
2.2 重要农艺性状相关基因的挖掘
参考基因组是挖掘与农艺性状紧密关联基因的基石。苦味是影响黄瓜商品性的重要农艺性状,在黄瓜参考基因组的基础上,结合黄瓜变异组图谱、传统的基因定位方法、生物化学与分子生物学等技术手段进行系统分析,发现9个控制黄瓜果实苦味物质葫芦素的合成基因以及2 个参与调控苦味物质合成的调控因子Bl和Bt[16-17,61],其中Bl调控叶片苦味、而Bt调控果实苦味[61]。
全基因组关联分析(GWAS)是研究基因组序列变异与目标性状关联程度、挖掘候选基因的重要方法。在葫芦科作物基因组测序的基础上,基于核心种质资源或自交系材料的重测序开展全基因组关联分析,挖掘到许多控制重要农艺性状的候选基因。Zhao 等[32]通过1 067 份甜瓜资源的全基因组关联分析,除鉴定到已报道的性别决定基因CmACS-7、果肉颜色基因CmOr、果皮颜色基因CmKFB和酸度基因CmPH外,还分别获得76、29 和99 个与产量、果实品质和外观性状相关的基因位点。基于146 份冬瓜核心种质资源的重测序和全基因组关联分析结果,Yan等[62]和Luo 等[63]进一步通过基于遗传图谱的基因定位,初步明确编码跨膜O-酰基转移酶和YABBY 转录因子的基因分别控制冬瓜果实表面蜡粉和种子籽型形成的候选基因。Du 等[64]在全基因组关联分析基础上,进一步通过基因定位推断一个编码AGAMOUS MADS-box 转录因子的MELO3C019694.2为决定甜瓜果实表面棱沟有无的候选基因。
3 展望
3.1 高质量参考基因组组装
葫芦科不同物种参考基因组的绘制,显著促进了该领域分子生物学研究水平。然而,前期的Roche 454、Sanger 和Illumina 测序平台的读长相对较短(100~150 bp),且依赖于遗传图谱辅助组装,高重复区域或着丝粒区域存在大量的gap 区域,限制了其在后续研究中的应用。随着测序技术的发展,三代测序读长显著提升(如Nanopore 读长可达150 kb),可以填补基因组中大片段的gap。近年来,基于PacBio HiFi、Hi-C 及Nanopore ultralong 测序技术,构建端粒到端粒(T2T)基因组逐渐成为研究热点。T2T 基因组具有高度准确性、连续性、完整性,有助于深入研究基因组中高重复序列区域,为研究着丝粒区域或未知高重复区域的变异特征提供了契机。在葫芦科作物中,西瓜[38]和苦瓜[47]T2T 参考基因组已有报道,其基因组的连续性和完整性得到显著提高。因此,对其他参考基因组质量相对不太完善的葫芦科作物,组装T2T 高质量参考基因组可能将为新功能基因鉴定和物种遗传变异分析奠定基础。
3.2 泛基因组研究
葫芦科作物同一物种内遗传变异丰富,单一或少数几个参考基因组不能完整呈现这些资源中的所有遗传变异,以单一参考基因组进行基因组学研究容易出现偏差或错误。构建来自多个个体的高质量泛基因组,不仅能在基因组水平上更全面地解析物种间的遗传变异,探明不同个体表型差异的遗传基础,而且可通过对多个物种、亚种间基因组比较分析,挖掘其特有基因和变异位点,为功能基因研究和全基因组设计育种奠定基础。目前,黄瓜、西瓜、甜瓜已完成多个参考基因组和大量核心资源基因组重测序,并利用全基因组关联分析挖掘了多个重要性状的关键候选基因。然而,其他葫芦科作物虽然组装了参考基因组,但是基因组深入分析与利用不足。因此,泛基因组构建及应用将成为今后葫芦科作物基因组学研究的热点。
猜你喜欢
小猕猴智力画刊(2022年9期)2022-11-04 02:32:02
中老年保健(2021年5期)2021-08-24 07:08:02
今日农业(2020年16期)2020-12-14 15:04:59
小学生学习指导(低年级)(2020年10期)2020-11-09 09:21:54
中国瓜菜(2020年6期)2020-07-08 09:47:18
热带农业科学(2018年1期)2018-07-04 11:33:02
中国瓜菜(2017年10期)2018-02-03 18:46:35
小学生导刊(2017年19期)2017-07-19 13:41:50
长江蔬菜·学术版(2017年1期)2017-04-11 07:41:26
中国蔬菜(2016年8期)2017-01-15 14:23:41
