
Ⅰ型神经纤维瘤病患者1例及其家系NF1基因突变检测
2023-06-07 06:28马盼盼康启超陈雪王兴田芯瑗郝胜菊惠玲徐福蓉张钏
中国皮肤性病学杂志 2023年6期
马盼盼,康启超,陈雪,王兴,田芯瑗,郝胜菊,惠玲,徐福蓉,张钏
Ⅰ型神经纤维瘤病(neurofibromatosis type1,NF1,OMIM: 162200)是一种由NF1基因突变导致的常染色体显性遗传病,成年先证者该病完全外显[1]。NF1通过影响来源于胚胎神经鞘的组织而导致多种临床表现[2]。该病主要临床表现包括牛奶咖啡斑、皮肤及皮下神经纤维瘤、中枢和周围神经系统的恶性肿瘤,其他不常见的临床表现包括学习障碍和骨骼系统异常(如:脊柱侧弯)等,并且其临床表型随着年龄的增加而增多和加重[3]。NF1的发病率约为1∶3 500,自发突变率为50%左右[4]。本文报告1例家族遗传性NF1患者,通过全外显子测序对该患者进行基因变异分析,对可疑变异位点进行Sanger测序验证,明确其致病基因变异位点,探讨其致病原因。
1 资料与方法
1.1临床资料 先证者来自于甘肃省妇幼保健院医学遗传中心。先证者男,33岁,自述10岁左右自己发现皮肤上有咖啡斑,近3年来出现手脚麻木、无力、脖子不能扭转,不能从事体力劳动。查体:胸前及腰间有多个牛奶咖啡斑(图1a),身体多处发现神经纤维瘤,先证者通过脊椎手术进行了部分神经纤维瘤的切除,症状有所缓解,脖子可以转动。诊断:NF1。先证者父亲也有多个牛奶咖啡斑,出现时间不详,背部有多个神经纤维瘤突起(图1b)。先证者妻子未发现牛奶咖啡斑,先证者与妻子结婚9年未自然怀孕,辅助生殖生育双胞胎男孩,现4岁,均有牛奶咖啡斑(图1c~1d)。该患者有家族史,家系图见图2。
1.2标本采集及基因组DNA提取 经先证者及其家属知情并签署基因检测知情同意书。分别抽取先证者及其家系外周静脉血各2~3 mL,EDTA抗凝。使用天根DNA提取试剂盒提取标本DNA,NanoDrop2000测定DNA的浓度均在50 ng/μL左右,OD260/280在1.8~2.0之间。本研究经本院伦理委员会([2021]GSFY伦审[65]号)批准。
1.3目标序列捕获、高通量测序及生物信息学分析
1.3.1样本基因组DNA片段化、末端修复及3′加“A” 利用酶切的方式将基因组DNA打断为200 bp左右的小片段DNA,末端修复和3′加“A”。
1.3.2接头连接及加标签 利用标准文库建库试剂盒对小片段DNA段进行Adapter接头连接,再加入Index标签,并通过Qubit 3.0 Fluorometer和Agilent 2100 Bioanalyzer system进行文库质检。
1.3.3目标区域捕获和高通量测序 生物素特异标记的探针与文库DNA进行杂交,链霉亲和素修饰的捕获磁珠捕获目的基因。采用Illumina Next Seq CN500测序仪进行测序。
1.3.4数据筛选与生物信息学分析 应用分析平台(北京迈基诺公司)进行致病基因筛选。应用ClinVar(https://www.ncbi.nlm.nih.gov/clinvar/)及人类基因突变数据库(Human Gene Mutation Database,HGMD)(http://www.hgmd.cf.ac.uk/ac/index.php)分析该位点变异。
1.4Sanger验证 使用在线Primer 3.0(http://primer3.ut.ee)软件设计引物,扩增引物序列:F 5′-GCAACAGAGGAAGACACCAT-3′,R:5′-CACAGTTTGACACAGGCAAT-3′。对候选致病基因NF1变异位点区域进行PCR扩增,Sanger测序验证。测序结果与NF1基因参考序列(NM_000267)进行序列比对。
2 结果
2.1全外显子基因检测结果 经全外显子测序数据分析,筛选到与先证者临床表型相关的变异:NF1基因c.889-2_889-1insTTTCTG位点杂合变异,该变异位于NF1基因内含子8上,即剪接位点第c.889-2_889-1位插入6个碱基TTTCTG,可能导致外显子的跳跃或内含子的滞留。
2.2Sanger测序验证结果 应用Sanger测序对先证者全外显子组测序结果进行验证。Sanger测序结果与全外显子组测序结果一致,先证者NF1基因c.889-2_889-1insTTTCTG位点杂合变异。同时对家系其他成员进行该位点Sanger测序,结果提示先证者父亲及其两个双胞胎儿子均存在NF1基因c.889-2_889-1insTTTCTG位点杂合变异,先证者之妻未见该位点变异(图3)。
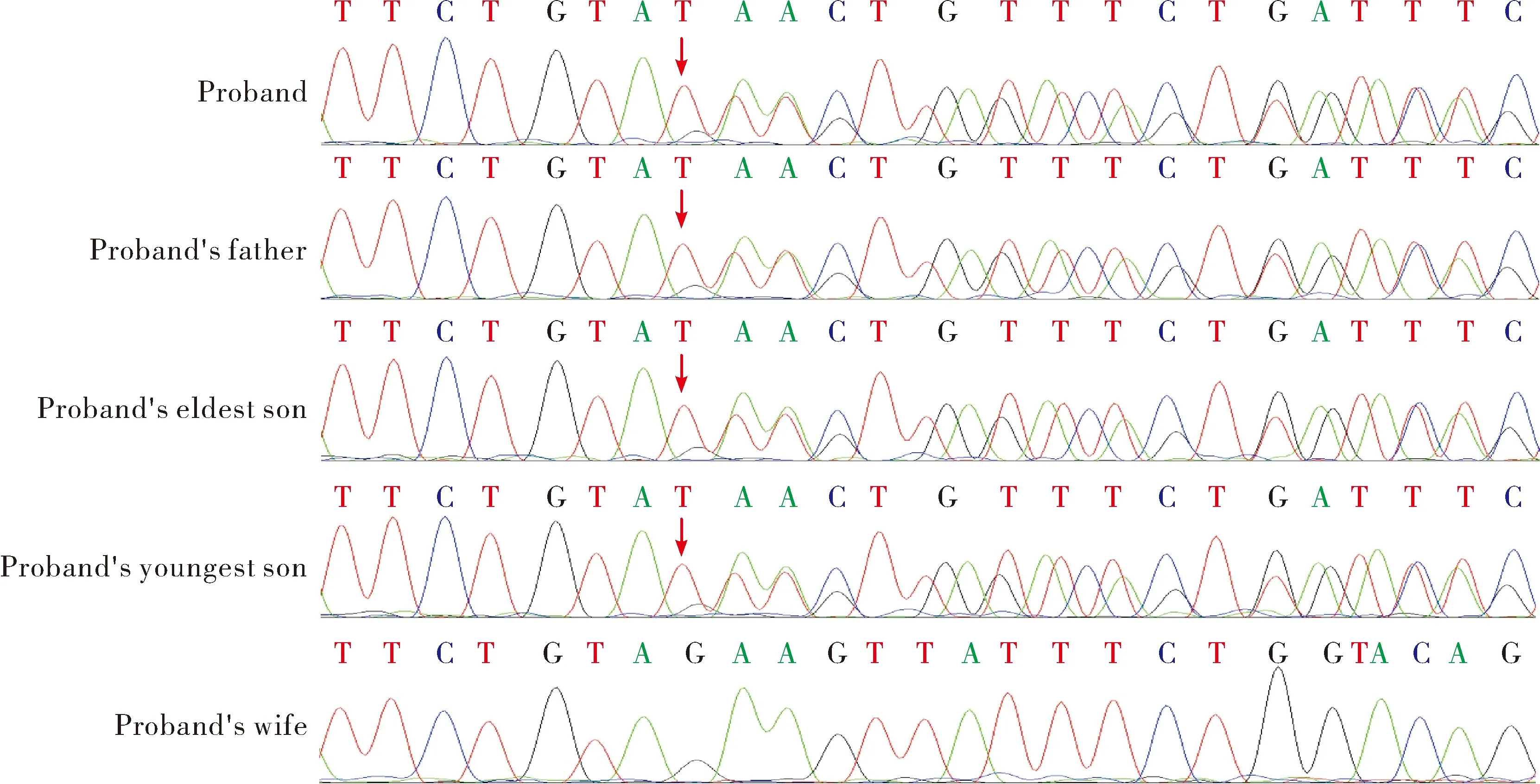
图3 先证者家系NF1基因变异位点Sanger测序图(红色箭头示变异位点)
2.3致病性分析结果 根据美国医学遗传学和基因组学学会(American College of Medical Genetics and Genomics,ACMG)2015年指南[5]对该变异位点进行致病性分析,c.889-2_889-1insTTTCTG变异为NF1基因剪接位点插入突变,该变异导致基因功能可能丧失,为超强致病证据(PVS1);在正常人群数据库中该变异频率小于0.000 5,为低频变异,为中等致病证据(PM2);该突变相关的疾病与先证者临床表型高度吻合,为支持证据(PP4)。因此该变异评级为致病性变异。文献数据库未有该位点的相关性报道,ClinVar及HGMD数据库均未收录该位点。
3 讨论
Ⅰ型神经纤维瘤病是由肿瘤抑制基因(NF1基因) 突变导致的。NF1基因位于17q11.2,全长350 kb。NF1基因的表达产物为神经纤维蛋白,由2 818个氨基酸组成,有抑制肿瘤的作用[6]。HGMD已报道的NF1突变类型目前已超过3 000种,突变类型包括缺失/插入突变、错义突变、无义突变、剪接突变、复杂重排等,尚未发现明确的突变热点。NF1的诊断主要依据1988年美国国立卫生研究院(National Institutes of Health,NIH)制定的Ⅰ型神经纤维瘤病的诊断标准。包括:①6个及以上的牛奶咖啡斑,青春期前直径大于5 mm,青春期后直径大于15 mm;②2个及以上任何类型的神经纤维瘤,或1个丛状神经纤维瘤;③腋窝或腹股沟雀斑;④视神经胶质瘤;⑤2个及以上虹膜错构瘤(Lisch 结节);⑥骨损害,如蝶骨翼发育不良,长骨皮质细线化,有或无假关节;⑦一级亲属关系中有符合上述标准的NF1。以上标准符合2条即可确诊NF1[7]。
本家系中先证者临床表型根据NF1诊断标准可以临床诊断为Ⅰ型神经纤维瘤病。但是在先证者未行基因检测的情况下,即进行辅助生殖,导致出生的双胞胎儿子也同样为NF1患者。因此,在做人工辅助生育之前,临床医生应仔细询问患者病史并进行相应的体格、皮肤检查;有临床遗传病家族史的夫妻做人工辅助生育之前应该先做相关基因检测。明确诊断后,再通过辅助生殖技术、胚胎植入前基因诊断和孕中期产前单基因病诊断技术来阻断遗传病患儿的出生。
NF1可出现多种临床表现,除常见的表型之外,文献报道NF1患者也有皮肤麻木、肢体麻木、乏力的表型[8],研究发现,剪切位点突变常与脊髓肿瘤相关[9]。本文对先证者全外显子进行检测,发现NF1基因8号内含子剪接位点杂合性插入突变c.889-2_889-1insTTTCTG,根据ACMG指南评级为致病性变异。Sanger测序检测到先证者、先证者父亲和先证者的双胞胎儿子均存在该位点杂合变异,先证者妻子未发现该变异。根据临床表型、基因检测结果和家系调查可以诊断此家系患者为Ⅰ型神经纤维瘤病,致病基因突变为NF1基因c.889-2_889-1insTTTCTG位点。
剪接突变是人类遗传病突变类型中常见的一种,其导致的结果有外显子的跳跃、内含子的滞留等。在哺乳动物基因组中,剪接位点为经典GT-AG组合的概率为98.71%[10-11]。正常情况下NF1基因c.889-5_895位点序列为tgtagAAGTTAT(小写为内含子,大写为外显子),发生突变后此段序列为tgtaTTTCTGgAAGTTAT(小写为内含子,大写为外显子,下划线标出为插入序列)。正常情况下剪接发生于c.889-2_889-1AG,9号外显子从c.889_890AA开始编码氨基酸。突变发生以后剪接只能发生于后面的c.890_891AG,9号外显子从c.892_893TT开始编码氨基酸,导致阅读框移位。由于实验条件限制,本研究尚未对NF1基因c.889-2_889-1insTTTCTG位点进行剪接影响分析,因此该突变对NF1基因mRNA剪接的影响还需进一步的研究。
Ⅰ型神经纤维瘤病的表型多种多样,一个家系中同样的突变会有不同的表型。虽然具体表现、进展速度和并发症的严重程度差别很大,但病情在个体一生中是逐渐进展的[12]。目前没有有效的治疗方法,临床治疗通常局限于监测和对症治疗,通常是手术治疗特定的并发症[13]。本家系中4人携带NF1基因c.889-2_889-1insTTTCTG位点突变,但是他们的临床表型轻重不一。先证者除了有明显的牛奶咖啡斑之外,全身多处有神经纤维瘤,进行手术部分切除。但是文献报道神经纤维瘤不易彻底剔除,容易复发,应定期检查[14]。相比较之下,先证者父亲也有多处牛奶咖啡斑,背部有神经纤维瘤状突起,但没有明显的神经症状。可能是因为神经纤维瘤长在不同部位影响不同部位的功能,从而表现出不同的表型,也有可能是常染色体显性遗传病所有的遗传早现现象导致的。先证者两个双胞胎儿子目前只是有牛奶咖啡斑,没有发现神经纤维瘤,可能是年龄尚小未表现出,应定期体检、随访。
总之,本研究对1例遗传性Ⅰ型神经纤维瘤病患者及其家系进行了表型和基因型的研究。NF1基因c.889-2_889-1insTTTCTG位点杂合变异为该家系的致病原因,且该位点为未见报道的新发变异,扩充了NF1基因突变谱,为Ⅰ型神经纤维瘤病患者的诊断提供了依据。对于辅助生殖的夫妇,一定要进行详细的病史问询和体格检查来判断是否有遗传病存在的可能,如果存在遗传病需明确诊断后再进行辅助生殖,通过胚胎植入前基因诊断和孕中期产前基因病诊断技术来阻止患儿的出生。
猜你喜欢
临床输血与检验(2022年3期)2022-06-22
内蒙古师范大学学报(自然科学汉文版)(2021年3期)2021-06-01
生物工程学报(2019年6期)2019-07-10
郑州大学学报(医学版)(2019年3期)2019-06-03
传染病信息(2019年2期)2019-05-17
生物学通报(2019年1期)2019-02-15
生物学通报(2018年12期)2018-10-10
广东海洋大学学报(2015年4期)2016-01-13
听力学及言语疾病杂志(2015年5期)2015-12-24
首都医科大学学报(2015年4期)2015-12-16
