
泛素连接酶与帕金森病发病机制中线粒体自噬和内质网应激的研究进展
2023-06-04 11:07梁宇任铭心王凯丽仲光尚叶明刘长青
沈阳医学院学报 2023年3期
梁宇,任铭心,王凯丽,仲光尚,叶明,刘长青*
(1. 蚌埠医学院临床医学院,安徽蚌埠 233000;2. 蚌埠医学院生命科学学院;3. 蚌埠医学院第一附属医院)
帕金森病(Parkinson's disease,PD)是仅次于阿尔兹海默症的第二大退行性疾病,多发于中老年,临床上可分为散发性PD 和家族性PD[1]。PD 的病理变化以中脑黑质的多巴胺能神经元缺失和路易小体(Lewy bodies,LBs)的形成为其主要病理特点[2]。其发病机制包括遗传和环境因素,分为LBs 内α-突触核蛋白(α-synuclein,α-syn)的聚集、泛素-蛋白酶体系统(ubiquitin proteasome system,UPS)损伤、内质网应激、线粒体异常及神经炎症等因素[3]。这些复杂的相互关系的发病机制和神经细胞程序性死亡是发病的必要途径。然而PD 的发病机制目前仍不清楚。UPS可以通过泛素修饰蛋白质发挥降解或调节作用,其损伤是上述PD 发病机制中众多诱因的基础。因此,本文就线粒体自噬和内质网应激这2种细胞器的泛素依赖型保护反应作一综述。
1 UPS简介
UPS 是ATP 依赖的高度特异性和选择性的蛋白水解系统,通过多个步骤降解错误折叠或聚集的蛋白质。UPS 可以调节细胞中80%的蛋白质的降解,几乎涉及细胞生理发育的所有方面,包括神经退行性疾病、肿瘤、免疫、病毒感染和细胞凋亡等[4]。
UPS 包括泛素(ubiquitin,Ub)、E1 泛素激活酶、E2 泛素结合酶、E3 泛素连接酶、26S 蛋白酶体和去泛素化酶。人体中有2 种E1 泛素激活酶(Uba1 和Uba6),约40 种E2 泛素结合酶和600 多种E3 泛素连接酶[5]。E3 泛素连接酶可分为3 种:HECT 型、RBR 型、RING 型(包括Ubox 型)[5]。不同类型的E3 泛素连接酶将Ub 传递到底物蛋白的方式不同[6]。其中大部分E3 是RING 型,可见E3 泛素连接酶功能作用的复杂性和多样性[7]。
Ub是UPS的核心部分,是由76个氨基酸残基组成的一种高度保守的小蛋白质(约8.6 KDa)[8]。Ub标记蛋白的水解反应比较复杂。首先,E1泛素激活酶以ATP 依赖的方式在泛素C 端的甘氨酸残基上激活泛素,而后者则通过硫酯键与E1 泛素激活酶的 Cys 残基结合,从而形成Ub-E1 复合体。E2 泛素结合酶随后也通过同样的硫酯键将Ub-E1激活的泛素转移到自身的Cys上,从而形成Ub-E2。接下来E3 泛素结合酶能够识别出Ub-E2 与底物蛋白,并将其直接或间接地转移到底物蛋白上,进而对底物蛋白进行Ub 修饰[9]。经过Ub 修饰的底物蛋白会在不同的生理学进程中起作用,这些过程由Ub 链来决定。底物的泛素化可以通过单个Ub 修饰分子(单一泛素化)、几个单个Ub 修饰分子(多泛素化)、Ub 修饰链(多聚泛素化)来调节[10]。Ub 有7 个赖氨酸残基(Lys6、Lys11、Lys27、Lys29、Lys33、Lys48 和Lys63)及其N 端(Met1),可共价连接形成泛素链,执行不同的生理功能[11],其中通过Lys48 连接的泛素链可以介导26S蛋白酶体对底物的降解,这是泛素化降解的主要链型[12]。
2 泛素连接酶与PD的相关性
600 多种E3 泛素连接酶在身体的各个部位表达,通过发挥不同的功能维持着生理环境的稳定。同样E3 泛素连接酶在调节神经退行性疾病中也具有至关重要的作用,通过调节线粒体自噬、内质网应激、神经炎症和细胞凋亡等影响神经系统疾病的发生进展[13-14]。α-syn 的异常聚集导致PD 的发生,其与线粒体自噬、内质网应激等均可被Parkin 等泛素连接酶调控,可见泛素连接酶的缺失或突变是PD 发病机制的基础。目前与线粒体自噬和内质网应激有关的主要E3 泛素连接酶有Parkin、 CHIP、 SIAH1、 FBXO7 和 HRD1 五种[15-16],而不同种类泛素连接酶发挥各自特异性的作用。
3 泛素连接酶与PD 发病机制中线粒体自噬的相关性
线粒体是一种高度动态的双膜细胞器,在真核细胞中发挥着广泛的功能。在活性氧、缺氧等外界环境刺激下可以引起线粒体的损害,从而引起线粒体的自噬。线粒体自噬是一种能够选择性地去除受损或多余线粒体的自噬过程,与家族性和散发性的PD有显著的病理相关性[17]。PARK6编码的蛋白激酶PINK1 及PARK2 编码的E3 泛素连接酶Parkin 被确定是早发性常染色体隐性遗传PD 的致病基因[18]。PINK1/Parkin 是泛素依赖型线粒体自噬的重要调节通路,PINK1 是Parkin 的上游因子,它能激活Parkin 的E3 泛素结合酶活性,PINK1/Parkin 突变会导致线粒体自噬功能异常,促进PD的发生和发展[19]。E3泛素连接酶FBXO7、CHIP 和SIAH1 也是PINK1 的下游底物,参与线粒体自噬。
3.1 PINK1/Parkin 介导的线粒体自噬 PINK1 和Parkin 分别是Ser/Thr 激酶和RBR 型E3 泛素连接酶,是泛素依赖型线粒体自噬途径的关键蛋白,其缺失可能导致PD。在正常的线粒体中,PINK1激酶以膜电位依赖性方式进入线粒体内膜。然后PINK1被线粒体内膜PARL等蛋白酶切割,这些被切割的PINK1 被逆向转移到胞质中,由蛋白酶体进行N 端规则降解[20]。所以,PINK1 在正常情况下的含量非常低,使其难以被检测到。在线粒体应激和去极化之后,PINK1 进入线粒体机制的通路被切断。这样PINK1 即在线粒体的外膜上聚集,从而激活Parkin。PINK1首先在Ser228位点进行自身磷酸化[21],激活后募集并在Parkin Ser65 位点磷酸活化Parkin,进而泛素化线粒体外膜蛋白OMM,介导线粒体自噬[22]。同时PINK1 也可以在Ser65 磷酸化Ub,进一步募集活化Parkin[23]。Ub 磷酸化和Parkin 是互相依赖的,线粒体上磷酸化Ub 的积累需要Parkin,而Parkin 的募集也取决于Ub的磷酸化[24]。
3.2 FBXO7 通过PINK1/Parkin 通路影响线粒体自噬 FBXO7 是Skp-Cullin-F-box(SCF)E3 泛素连接酶的一段,由PARK15 编码,其突变导致早发性常染色体隐性遗传的PD[25]。正常情况下,FBXO7 主要位于细胞核中,氧化应激会使FBXO7 转运并聚集到线粒体,形成的聚集体会损害线粒体功能[25]。另一方面,FBXO7 可作为辅助蛋白通过与PINK1 和Parkin 相互作用参与线粒体的质量控制[26]。FBXO7 可促进Parkin 转移到去极化的线粒体上,而FBXO7 又需要PINK1 才能转移到线粒体中[27]。在哺乳动物中,Parkin 以依赖FBXO7 的方式促进线粒体自噬,沉默FBXO7会抑制Parkin 的寡集和线粒体自噬[27]。FBXO7 基因突变导致的临床症状类似于PINK1 和Parkin 基因突变所产生的症状[28],但是FBXO7 突变患者通常不会在受影响的脑区产生α-syn,这是其独有的病理特征[25]。
3.3 CHIP 与Parkin 协同介导线粒体自噬 CHIP(Hsp70 相互作用蛋白)是U-box 型E3 泛素连接酶,自发现以来,CHIP 已经被证明与神经系统疾病的发生发展有关,包括卒中、脑出血,阿尔兹海默症、PD等[29]。其中CHIP影响PD中线粒体自噬和α-syn 的发生发展。有研究表明,CHIP 可以快速定位到受损伤的线粒体,并通过线粒体自噬发挥作用[30]。在果蝇中,CHIP在PINK1信号传导的下游发挥作用,当Parkin 沉默表达时,CHIP 的过表达可以挽救线粒体自噬[31]。Parkin 和CHIP 具有协同性,它们可能有共同的作用底物Miro、Gdap-1、Dnm3 和Drp1 以介导线粒体自噬[32]。然而具体机制和CHIP 介导线粒体自噬有没有其他途径还需要进一步研究。
3.4 PINK1-synphilin-1-SIAH1 介导线粒体自噬α-syn 相关蛋白synphilin-1 存在于PD 患者的LBs中[33],常与α-syn 作为LBs 的标志物。虽然synphilin-1 定位于突触小泡附近,但其功能仍然不清楚。Parkin 的IBR 结构域与E2 结合酶家族的UcbH7 和UcbH8 蛋白相互作用以促进synphilin-1泛素化[34],α-syn、synphilin-1 和Parkin 三者共表达导致出现LBs 内泛素阳性[35]。研究发现,PINK1-synphilin-1-SIAH1 是一种不依赖与Parkin的线粒体自噬途径,抑制Parkin 表达对PINK1-synphilin-1-SIAH1 介导的线粒体自噬没有影响[36]。其机制是PINK1 将synphilin-1 募集到线粒体,后者再募集SIAH1,进而泛素化线粒体蛋白OMM,介导LC3 募集并运送至溶酶体进行线粒体自噬。
4 泛素连接酶与PD 发病机制中内质网应激的相关性
内质网控制着约1/3 蛋白质的合成、折叠和转导后修饰,还负责脂质和类固醇激素的合成,是Ca2+的主要贮存位点[37]。内质网稳态主要受未折叠蛋白质反应(unfolded protein response,UPR)调节,内质网腔内Ca2+浓度改变,发生氧化应激或者合成运输的蛋白质发生了一些突变时,将导致错误折叠的蛋白质在内质网中积累,进而引发内质网应激,并激活UPR[38]。错误折叠的蛋白质也可以被内质网相关降解途径(ER-associated degradation,ERAD)清除,ERAD 是指将错误折叠的蛋白质运出内质网后被UPS 系统降解的过程。在哺乳动物中,UPR 由内质网膜上IRE1、PERK 和ATF6 信号通路介导激活,当内质网应激持续时间过长时,细胞功能将会发生障碍,最终导致细胞凋亡,而这是通过CHOP、JNK 和Caspase12 等途径介导的[39]。越来越多的研究表明,内质网应激与PD 的联系存在于遗传和各种非生物因素的PD 模型中[40]。内质网内的α-syn 聚集在体内体外通过PERK 途径诱导了UPR 激活[41]。当蛋白酶体功能受损时,内质网中积累的错误折叠蛋白不能被有效降解而导致神经元变性[42]。
4.1 Parkin 调节内质网-线粒体的相互作用 Imai等[43]早在2000年就发现Parkin可以抑制UPR诱导的细胞凋亡,这是因为其具有E3 泛素连接酶活性。Parkin 可以被UPR 的PERK/ATF4 信号诱导上调[44],然后作为一种参与了ERAD 的E3 泛素连接酶,针对性地泛素化降解内质网腔内的未折叠蛋白质,保护神经元免受内质网应激诱导的细胞凋亡[45]。Imai 等[46]发现,在AR-JP 中,Parkin 抑制Parkin 相关的内皮素受体样受体(Pael-R)积累诱导的内质网应激,Pael-R 是ER 应激诱导因子和家族性PD的致病因素;并且CHIP可正向调节Parkin的E3 活性,增强其抑制Pael-R 诱导的细胞凋亡的能力[46]。另有研究发现,C/EBP 同源蛋白CHOP是内质网应激引发细胞凋亡的调节因子,其可被Parkin 泛素化并被26S 蛋白酶体降解[47]。Parkin 也可以E3 泛素连接酶活性抑制JNK 信号,抑制细胞凋亡[48]。可见Parkin 在内质网应激中发挥着重要的神经保护功能。
此外,Parkin在内质网和线粒体中的作用可以相互联系并彼此影响。内质网与线粒体的接触位点称为线粒体相关膜(MAM),Ca2+从内质网转移到线粒体是MAM 的基本功能[49]。PINK1和Parkin也表达于MAM 中,最近研究发现,在果蝇PINK1/Parkin 突变体中,损伤的线粒体会通过激活UPR 的Perk 途径产生内质网应激信号[50]。还发现PINK1/Parkin磷酸泛素化MFN2,促进线粒体自噬和线粒体与内质网分开[51]。MFN2是MAM中的关键组成部分,敲除MFN2 可以抑制内质网-线粒体相互作用[52]。然而,有与之相反的研究表明,PINK1/Parkin 突变或抑制表达导致内质网和线粒体的相互作用减少[53-54]。这2 种矛盾的学说表明PINK1/Parkin 在内质网-线粒体中具有重要作用,但仍不完全清楚。
4.2 HRD1 通过降解Pael-R 抑制内质网应激 HMG-CoA还原酶降解蛋白1(HRD1)包含有E3 的环指结构域,在黑质致密部的多巴胺能神经元中广泛表达,主要定位于内质网膜上。其与Parkin均在ERAD 中发挥保护作用[55],HRD1被内质网应激的IRE1 和ATF6 信号上调,进而泛素降解错误折叠的蛋白质[56]。上调的HRD1 可以泛素化并降解EIF2α 和IRE1α,抑制PERK-EIF2α-ATF4-CHOP 和IRE1α-p38 信号通路,减少细胞凋亡[57-58]。同时HRD1 与Parkin 有相同的作用底物Pael-R,HRD1 可以介导Pael-R 的泛素化和降解,抑制其诱导的神经细胞凋亡[59]。而且当HRD1 抑制表达时,Parkin 表达上调;Parkin 抑制表达时,HRD1 表达水平不变[60]。所以Parkin 可以通过补偿机制缓解HRD1 突变导致的内质网应激和细胞凋亡。
4.3 CHIP 维持内质网应激诱导的适应反应 有研究发现,过表达的CHIP 抑制了由内质网应激诱导的CHOP 和p53 的上调,减弱了内质网应激诱导的凋亡反应[61]。同时CHIP 不能影响UPR 诱导的BiP/GRP78 上调,维持了内质网应激的适应反应[61]。内质网伴侣BiP/GRP78 是PD 中UPR 启动的重要蛋白。在未折叠蛋白的压力下,BiP/GRP78 会与PERK、IRE1 和ATF6 解离,进而发挥减弱翻译,促进蛋白质正确折叠等保护功能[62]。而且CHIP 也可泛素化IRE1,促进IRE1/TRAF2 复合物的形成,拮抗内质网应激诱导的衰老过程[63]。
E3 泛素连接酶参与调控线粒体自噬和内质网应激的作用机制见图1。
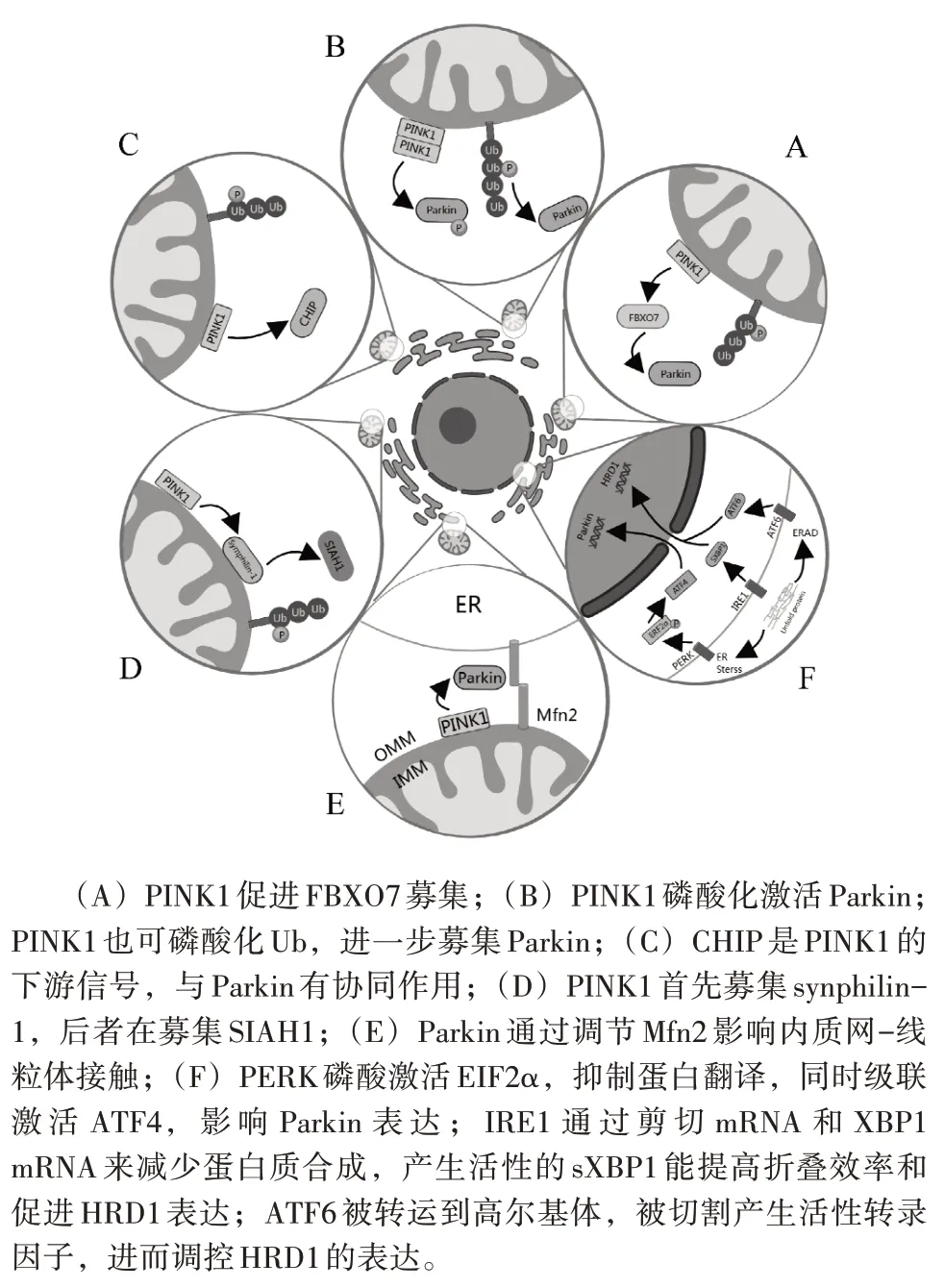
图1 E3泛素连接酶参与调控线粒体自噬和内质网应激
5 小结与展望
UPS 途径泛素降解系统近年来被广泛关注,其各种组成结构和功能均逐渐明确,冷冻电镜技术的发展将难以解密的26S 蛋白酶体结构得以窥视。但是泛素连接酶种类繁多,以Parkin 为代表研究较多的泛素连接酶,仍有很多机制需要进一步研究。Parkin通过调控线粒体自噬发挥保护和凋亡的机制如何阐明,什么原因导致两种不同的自噬结果;Parkin 在内质网-线粒体串扰中为什么具有两种相反的作用;这些问题仍有待后续进一步研究。不同的泛素连接酶,在某些方面也许有同样的功能。Parkin、FBXO7、CHIP 和SIAH1 具有神经保护作用,这是通过调节线粒体自噬来实现的;Parkin、HRD1 和CHIP 可抑制内质网应激介导的神经细胞凋亡。然而E3 泛素连接酶SIAH1 在PD 的发生发展中,与其它泛素连接酶不同,具有双重效应,SIAH 促进线粒体自噬的同时,也促进α-syn的单泛素化聚集。总之,这些E3泛素连接酶通过影响线粒体自噬和内质网应激途径,促进或者抑制PD 的发病过程,为后续研究UPS途径治疗PD提供了有价值的治疗线索。
猜你喜欢
解放军医学杂志(2021年12期)2022-01-18
现代临床医学(2021年1期)2021-01-26
华东师范大学学报(自然科学版)(2018年2期)2018-05-14
科学中国人(2017年36期)2017-06-09
安徽医科大学学报(2016年12期)2017-01-15
中国病理生理杂志(2015年8期)2015-12-21
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15
中国医学科学院学报(2015年5期)2015-03-01
中国当代医药(2015年33期)2015-03-01
现代检验医学杂志(2015年2期)2015-02-06
