
PI3K/AKT通过激活糖酵解促进巨噬细胞M1极化
2023-05-28 13:25马佳睿徐芳芷左惠演满永张曦月窦琳唐蔚青刘进黄秀清黎健
中国心血管杂志 2023年2期
马佳睿 徐芳芷 左惠演 满永 张曦月 窦琳 唐蔚青 刘进 黄秀清 黎健
100730 北京医院 国家卫生健康委北京老年医学研究所 国家卫生健康委北京老年医学重点实验室 国家老年医学中心 中国医学科学院老年医学研究院(马佳睿、徐芳芷、左惠演、满永、张曦月、窦琳、唐蔚青、刘进、黄秀清、黎健);100875 北京师范大学生命科学学院抗性基因资源与分子发育北京市重点实验室(刘进)
目前,我国心血管病发病率持续增高,心血管病死亡占城乡居民总死亡原因的首位[1]。巨噬细胞是天然免疫的关键群体,正常情况下巨噬细胞呈静息状态,当机体受到不同刺激后巨噬细胞可转化为M1经典活化型或M2交替活化型[2]。巨噬细胞的M1极化及所介导的炎症反应除发挥抗感染作用外,也与多种心血管病发病机制关系密切,如动脉粥样硬化、冠心病和心肌缺血等[2-3]。鉴于巨噬细胞在健康和疾病中的重要性,阐明巨噬细胞M1极化的机制具有重要意义。
早期研究中,对巨噬细胞M1极化的调控多集中于各种转录因子,如核因子κB(nuclear factor kappa B,NF-κB)、STATs和KLFs家族成员等对炎症因子的调控[4]。此外,MAPKs家族成员如P38、细胞外调节蛋白激酶(extracellular regulated protein kinases,ERK)等,也参与到M1极化调控中[5-7]。近期研究发现,糖代谢重编程可能对巨噬细胞极化十分重要[8-9]。诺贝尔奖得主Otto Warburg发现,与大部分细胞在有氧环境中优先通过氧化磷酸化供能的方式不同,肿瘤细胞即使在有氧条件下,也优先利用糖酵解来获取大部分能量,这种糖代谢方式的改变即称为Warburg效应[8-9]。Warburg效应最初被认为仅在肿瘤细胞中发生,近期的研究结果发现M1极化的巨噬细胞也出现与肿瘤细胞相似的Warburg效应[10]。使用己糖激酶抑制剂2-脱氧-D-葡萄糖(2-Deoxy-D-Glucose,2-DG)抑制糖酵解过程,可以抑制巨噬细胞M1极化过程中特征性炎症因子的产生[11],表明糖酵解在巨噬细胞极化及炎症发生中起重要的调控作用。
尽管学术界已认识到糖代谢转化与巨噬细胞命运密切相关,但其调控机制仍有许多未知之处。磷脂酰肌醇3激酶/蛋白激酶B(phosphatidylinositol 3 kinase/protein kinase B,PI3K/AKT)信号通路是机体内重要的调控通路之一,可由胰岛素、多种生长和存活因子激活,并参与调控了众多生物学过程如凋亡、存活调控和葡萄糖转运等。近期PI3K/AKT信号通路在糖代谢重编程中的作用备受关注,然而在巨噬细胞中,PI3K/AKT信号通路是否可以通过调控糖代谢重编程,进而影响其M1极化过程,目前研究较少。本研究以脂多糖(lipopolysaccharide,LPS)诱导建立巨噬细胞M1极化模型,从代谢重编程的角度探讨PI3K/AKT通路在M1极化中的作用。
1 材料与方法
1.1 材料
细胞总RNA提取试剂Trizol和H-DMEM培养基均购自Invitrogen公司;胎牛血清购自Gibco公司;LPS、2-DG及LY29400购自Sigma公司;反转录酶MMLV购自NEB公司;Q-PCR试剂盒SYRB Green、dNTP(10 mM)和通用引物oligo dT均购自TaKaRa公司;p-mTOR、mTOR、p-p38、p38、p-ERK、ERK、p-AKT、AKT、actin、p-p65、p65和IkB抗体购自CST公司;一抗稀释液购自Invitrogen公司;HRP标记山羊抗兔二抗购自中杉金桥公司;化学发光试剂盒ECL购自Millipore公司;Seahorse XF糖酵解速率测定试剂盒购自Agilent公司。
1.2 RAW264.7细胞的培养及药物处理
小鼠巨噬细胞株RAW264.7购自中国医学科学院细胞库。细胞使用H-DMEM培养基(含10%胎牛血清、1%双抗)于37℃、5%CO2恒温培养箱(Thermo公司)内培养。使用1 μg/ml LPS于无血清培养基中处理细胞6 h[12];使用2-DG抑制糖酵解[13]时,将5 mM 2-DG与LPS共处理6 h;使用LY294002抑制PI3K/AKT[14]时,将20 μM LY294002预处理细胞18 h后,加入LPS共同处理6 h。
1.3 糖酵解水平的检测
使用Seahorse XF糖酵解速率测定试剂盒(103344-100)测定细胞外酸化率(extracellular acidification rate,ECAR),将细胞接种在XF24微孔板中,并进行相应的药物处理。根据试剂盒说明,在无CO2培养箱中培养1 h,在3次基线ECAR测量后,依次向细胞添加Rot/AA(0.5 μmol/L)和2-DG(50 mmol/L),并使用Seahorse-XF 24细胞外通量分析仪(Seahorse Bioscient)收集数据及进行分析。
1.4 炎症因子和糖酵解相关基因mRNA水平检测
使用Trizol试剂提取细胞总RNA,使用MMLV反转录合成cDNA。按照SYBR Green说明书配制PCR反应液,使用Thermo公司荧光定量PCR仪器检测收集数据。采用2-ΔΔCt法处理所得数据,计算各个基因相对表达量。PCR引物序列如表1所示。

表1 q-PCR引物序列
1.5 Western blot检测
收集处理完毕的细胞,使用细胞裂解液RIPA(索莱宝公司)提取总蛋白,取15 μg总蛋白以12% SDS-PAGE电泳分离蛋白,300 mA恒流转印PVDF膜(Milliproe公司),于10%脱脂牛奶(配制于TBST)室温孵育2 h,用1:1 000一抗(配制于一抗稀释液)于4℃孵育过夜,TBST洗膜3次每次15 min;使用1:5 000 HRP标记山羊抗兔二抗(配制于TBST)室温孵育2 h;TBST洗膜3次每次15 min;使用化学发光液检测条带。
1.6 统计学方法
2 结果
2.1 LPS诱导的巨噬细胞发生M1极化
使用1 μg/ml LPS处理RAW264.7细胞6 h,检测巨噬细胞MAPK和NF-κB信号通路极化及其M1极化标志物的mRNA水平。结果显示,与对照组(1 μl/ml生理盐水处理)相比,LPS处理组细胞中,MAPK(图1A、1B)和NF-κB信号通路激活(图1C、1D)。即p-p38和p-ERK水平升高;p-p65水平升高,IκB水平降低;M1极化标志物iNOS(图1E)及TNF-α、IL-6和IL-1β(图1F)mRNA水平升高。表明LPS诱导的巨噬细胞M1极化模型构建成功。
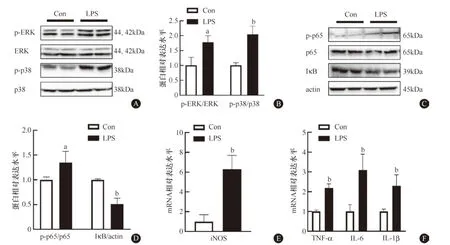
A:Western blot检测MAPK信号通路(n=3);B:MAPK信号通路的统计结果;C:Western blot检测NF-κB信号通路(n=3);D:NF-κB信号通路的统计结果;E、F:q-PCR检测M1极化标记物(n=6)。与对照组比较,aP<0.05,bP<0.01
2.2 糖酵解调控LPS诱导的巨噬细胞M1极化过程
为检测巨噬细胞M1极化模型中糖代谢重编程的情况,我们使用Seahorse XF糖酵解速率测定试剂盒检测LPS处理后细胞糖酵解水平,ECAR值显示LPS引起细胞体外酸化增加(图2A),q-PCR结果显示葡萄糖酵解相关基因(Glut1、HK2、Idha和Aldoa)mRNA水平升高(图2B),表明LPS诱导的巨噬细胞发生M1极化模型中,细胞发生明显的糖代谢重编程。
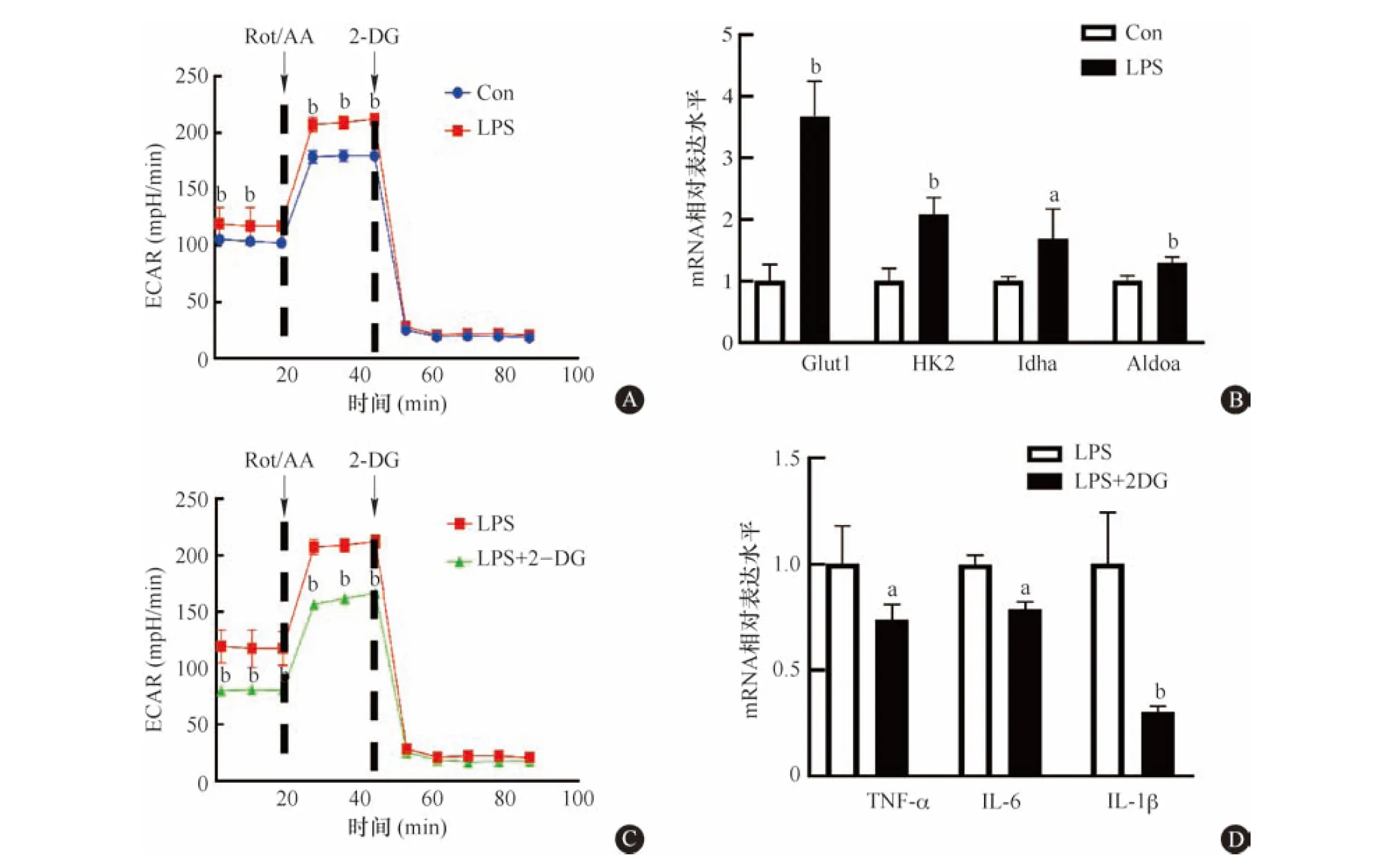
A:Seahorse XF糖酵解速率测定试剂盒检测LPS处理后糖酵解水平(n=5);B:q-PCR检测糖酵解相关基因mRNA表达水平(n=6);C:Seahorse XF糖酵解速率测定试剂盒检测2-DG处理后糖酵解水平(n=5);D:q-PCR检测2-DG处理后M1极化标记物(n=6)。与LPS组比较,aP<0.05,bP<0.01
为验证糖酵解在巨噬细胞M1极化中的作用,我们使用己糖激酶抑制剂2-DG抑制LPS诱导的细胞糖酵解过程。结果显示,2-DG显著抑制LPS诱导的ECAR值升高(图2C)。随着2-DG对糖酵解的抑制,巨噬细胞M1极化相关因子TNF-α、IL-6和IL-1β的mRNA水平也显著下降(图2D)。表明糖酵解调控LPS诱导的巨噬细胞M1极化过程。
2.3 抑制PI3K/AKT信号通路逆转LPS诱导的小鼠巨噬细胞糖代谢重编程及M1极化
为验证PI3K/AKT信号通路在LPS诱导的小鼠巨噬细胞糖代谢重编程及M1极化中的作用,我们首先检测了LPS处理后PI3K/AKT的活化。结果显示,LPS显著增加了p-AKT的水平(图3A、3B)。进一步使用LY294002抑制PI3K/AKT激活。Western blot结果显示,LY294002显著抑制p-AKT的水平(图3C、3D)。接着使用Seahorse XF糖酵解速率测定试剂盒检测PI3K/AKT抑制对LPS诱导的细胞糖酵解水平的影响,ECAR值显示PI3K/AKT抑制显著降低了LPS引起细胞糖酵解水平升高(图3E)。伴随着PI3K/AKT活性的抑制,葡萄糖酵解相关基因(Glut1、HK2、Idha和Aldoa)(图3F)和M1极化相关因子iNOS、TNF-α、IL-6和IL-1β的mRNA水平也显著降低(图3G、3H),表明抑制PI3K/AKT信号通路可逆转LPS诱导的小鼠巨噬细胞糖代谢重编程和M1极化。
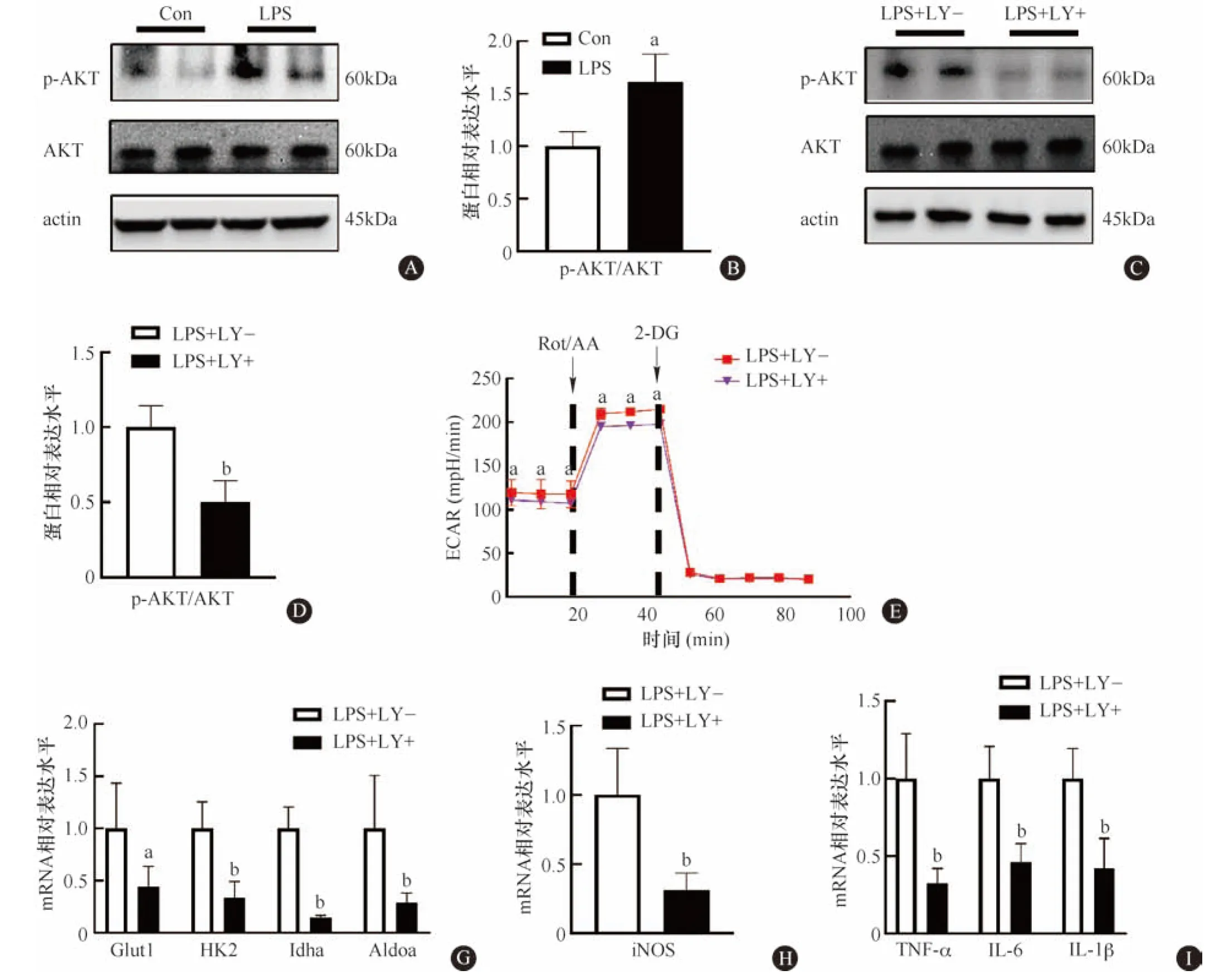
A、C:Western blot检测AKT激活(n=3);B、D:AKT激活的统计图;E:Seahorse XF糖酵解速率测定试剂盒检测LY294002处理后糖酵解水平(n=5);F:q-PCR检测LY294002处理后糖酵解相关基因mRNA表达水平(n=6);G、H:q-PCR检测LY294002处理后M1极化标记物(n=6)。与对照组或LPS+ LY294002-组比较,aP<0.05,bP<0.01
2.4 PI3K/AKT可能通过对mTOR通路的调节参与巨噬细胞代谢重编程和M1极化的调控
mTOR/HIF-1α是LPS诱导糖酵解中的核心调控通路,我们进一步检测PI3K/AKT信号是否通过对mTOR/HIF-1α通路的调节进而影响糖酵解和M1极化过程。与以往结果一致,LPS处理后mTOR信号迅速激活,表现为p-mTOR水平及HIF-1α蛋白水平升高(图4A~C)。进一步使用LY294002检测PI3K/AKT活性对mTOR信号通路的影响。Western blot结果显示,LY294002抑制p-AKT的水平后,除抑制NF-κB信号通路的激活外(图4G、4H),也显著降低p-mTOR和HIF-1α水平(图4D~F)。表明PI3K/AKT信号通路可能是通过mTOR信号参与LPS诱导的糖代谢重编程和M1极化的调控。
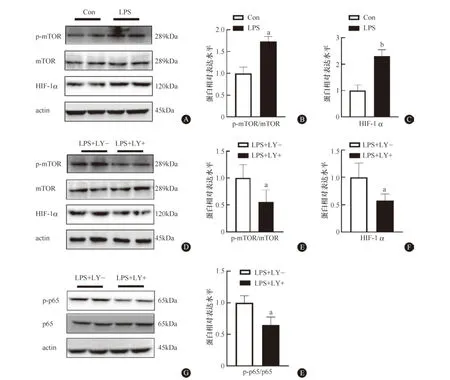
A、D:Western印迹检测mTOR的激活及HIF-1α的表达(n=3);B、E:mTOR激活的统计图;C、F:HIF-1α的统计图。与Con组或LPS+LY-组比较,aP<0.05,bP<0.01
3 讨论
在本实验中,我们初步证实了PI3K/AKT信号通过激活mTOR/HIF-1α信号轴,调节糖酵解,进而影响巨噬细胞M1极化进程。本研究从代谢重编程的角度阐述巨噬细胞M1极化中的机制,扩展了对巨噬细胞M1极化调控机制的认识。
巨噬细胞M1极化过程中产生的大量致炎因子,是机体对抗感染的重要武器,也与多种急慢性疾病的发生发展密切相关[2]。早期研究显示,转录水平的调控对巨噬细胞极化表型十分重要,目前已经鉴定出多个转录调节因子[4]。NF-κB是一种重要的转录因子,在LPS诱导的M1极化中,很多M1极化基因如iNOS、MCP-1等的启动子区域都包含NF-κB的结合位点。LPS结合其配体后,经由Toll样受体4激活NF-κB信号,从而促进多种M1极化相关的细胞因子产生[4]。此外,MAPKs家族成员介导的炎症因子产生也是促进巨噬细胞M1极化的重要途径[5-7]。本研究以LPS处理巨噬细胞,发现多个转录通路激活,M1极化标志物升高,表明模型构建成功。
近年来,代谢重编程在巨噬细胞表型转换中的作用越发显著[15]。1970年,研究者在LPS刺激的M1型极化巨噬细胞中发现了供能方式的改变,出现与肿瘤细胞相似的Warburg效应,即以糖酵解代替氧化磷酸化作为细胞主要的供能方式[10]。随后研究显示,糖代谢功能方式的转换不仅可以为巨噬细胞快速供给能量,还可以为M1极化过程中快速大量炎症因子产生过程提供所需的大量底物,是巨噬细胞M1极化的新干预靶点[8]。Seahorse XF糖酵解速率测定试剂盒是一种新兴的可准确可靠地评估糖酵解通量关键参数的方法。XF仪器直接实时测量细胞外酸化率ECAR值,反映糖酵解压力的变化。与以往研究结果一致,在LPS诱导的M1极化模型中,q-PCR结果显示糖酵解相关基因表达水平升高,Seahorse实验结果显示LPS诱导糖酵解水平升高,表明M1极化过程中确实发生糖代谢重编程。而使用己糖激酶抑制剂2-DG抑制糖酵解过程,抑制了巨噬细胞M1相关炎症因子的产生,表明这种代谢方式改变在巨噬细胞促炎表型转化中起重要作用。
在巨噬细胞糖代谢重编程中,mTOR/HIF-1α通路是调控巨噬细胞糖酵解过程的重要机制。mTOR复合物作为调控细胞代谢的核心信号具有重要的地位,在细胞内有两种不同蛋白复合物,称为mTORC1和mTORC2[16]。研究显示,mTORC1是调节糖酵解过程的核心蛋白复合物,由mTOR、Raptor和mLST8组成[17]。M1型巨噬细胞极化时,LPS激活mTOR,通过促进含有5’-末端寡嘧啶信号的mRNA翻译,增加HIF-1α的表达。HIF-1α是调控糖酵解代谢的关键蛋白[18],它能上调糖酵解途径中的关键酶——HK和Glut的表达[17]。我们的实验结果也显示了LPS可以诱导mTOR/HIF-1α轴的激活。
PI3K/AKT信号通路作为机体内重要的调控通路之一,可被多种刺激因素激活,在巨噬细胞M1极化过程中发挥重要作用。本研究显示,在LPS诱导的M1极化模型中,AKT活性显著升高,使用LY294002抑制PI3K/AKT活性,显著抑制LPS引起的糖酵解和M1极化过程,表明PI3K/AKT参与了LPS诱导的糖酵解的调控。以往研究提示,PI3K/AKT信号是mTOR通路的重要上游调控者[19],如PI3K/AKT激活mTOR信号促进巨噬细胞泡沫化的产生[20],我们进一步检测PI3K/AKT抑制对mTOR/HIF-1α信号通路的影响,以明确PI3K/AKT信号通路是否可通过调控mTOR/HIF-1α信号通路参与糖代谢重编程。正如我们所预期,PI3K/AKT抑制降低了mTOR/HIF-1α轴的激活以及相应的糖酵解过程。表明PI3K/AKT很可能是通过对mTOR/HIF-1α的调控参与LPS诱导糖酵解过程,进而参与M1极化的调控。
总之,巨噬细胞M1极化过程与多种心血管病发病机制及进程密切相关[21],我们的研究证实了PI3K/AKT信号通路在巨噬细胞糖酵解和M1极化中的重要调控作用,从代谢重编程的角度丰富了M1极化调控机制网络,这些研究结果将为心血管病的防治提供新的线索和思路。
利益冲突:无
猜你喜欢
中南医学科学杂志(2022年6期)2022-12-04
现代财经-天津财经大学学报(2022年5期)2022-06-01
少先队活动(2021年2期)2021-03-29
汽车维修与保养(2021年8期)2021-02-16
学生天地(2020年17期)2020-08-25
数学大王·低年级(2020年3期)2020-03-12
中国循证心血管医学杂志(2020年11期)2020-01-08
电子测试(2017年15期)2017-12-18
中华老年口腔医学杂志(2016年4期)2017-01-15
中国医学装备(2016年6期)2016-12-01
