
基于DNA 条形码及SSR 技术鉴定风藤和山蒟
2023-05-25 07:19:08严卓彦夏瑛瑛
现代中药研究与实践 2023年2期
严卓彦,夏瑛瑛,崔 洁,李 彬
[舟山市食品药品检验检测研究院(国家海洋食品质量检验检测中心),浙江 舟山 316012]
海风藤是我国传统的祛风湿药,自《名医别录》起,历代本草著作均有记载,具有悠久的应用历史[1]。从1985 年版至2020 年版《中国药典》[2]一直规定海风藤为胡椒科植物风藤Piper kadsura的干燥茎藤。但全国各地海风藤同名异物情况较多,容易混淆,文献记载的就有9 科19 种[3]。其中,山蒟最是频繁地被当作海风藤使用。《福建药物志》和《福建植物志》认为福建海风藤原植物主要是胡椒属植物山蒟Piper hancei及风藤[4]。孟静等[5]通过查阅古代本草、医籍,结合近现代文献资料,对海风藤药材名称、基原、产地及采收加工进行考证,也证实宋代之后海风藤药材使用品种多为山蒟和风藤。基于《中国药典》中海风藤原植物的规定与实际海风藤所用原植物存在偏差,研究风藤与其近缘物种山蒟之间的鉴别具有重要的实际价值。
山蒟为风藤同科同属植物,它的干燥茎藤以浙海风藤为名收载于2015年版 《浙江省中药炮制规范》[6]。在《浙江省中药炮制规范》中,山蒟与风藤的性状描述基本一致,且鉴别、检查、浸出物同2020 年版《中国药典》中“海风藤”项下规定。孙绍美等[7]对风藤及其同属植物的药效学评价和急性毒性研究,也证实山蒟具有风藤相同的药理活性及相似的活性强度。在分子水平上,刘艳菊等[8]对风藤和山蒟的基因组进行RAPD 鉴别研究,结论是两者的DNA 指纹图谱具有一定的相似性,可以确定两者的质量标准相似。风藤和山蒟在性状、化学成分和基因组上的相似程度均较高。
本研究基于DNA 条形码和简单重复序列SSR 分子标记技术,对来自全国各地的31 批风藤和山蒟药材植物和不同医药公司生产的药材饮片进行分析,旨在建立一种快速鉴别风藤与山蒟的方法,为中药材海风藤的种质资源鉴定提供一定的分子依据。
1 材料与仪器
1.1 材料
研究的样本信息见表1,其中有采摘自舟山的风藤和山蒟植物8 株,由浙江省食品药品检验检测研究院的郭增喜主任药师进行形态鉴别。来自四川、湖北、安徽的海风藤药材5 批,其余17 批次样品来自浙江省不同药材公司生产的海风藤和浙海风藤饮片,所有药材和饮片均由本院郑国平主任中药师进行形态鉴别。

表1 31 批海风藤山蒟样品的编号及来源信息Tab.1 Serial number and source of 31 batches of materials
1.2 仪器和试剂
BioRad My Cycler 型PCR 扩增仪、BioRad Mini-sub型水平电泳仪、Gel DocXR 型凝胶成像系统(美国伯乐公司);Termo nanodrop one 型微紫外/可见分光光度计、Thermo Lgend Micro 17 型离心机 (美国热电公司)。
2×CTAB 缓冲液:2% CTAB(十六烷基三甲基溴化铵),1%β-巯基乙醇(使用时现加入),1.4 moL NaCl,20 mmoL EDTA,100 mmoL Tris-HCl (pH = 8.0)。
Taq Plus DNA 聚合酶(B600090)、10X PCR Buffer(B600017)、dNTP(B500056),均购自生工生物工程上海股份有限公司。
2 方法
2.1 DNA 提取
将植物原株的风干根茎在75%酒精中浸泡1 min消毒后用无菌水冲洗,剪碎。将切片药材直接放入75%酒精中浸泡1 min 消毒后用无菌水冲洗。然后液氮研磨至细小颗粒,收集样本颗粒,在-70℃冰箱中保存。采用改进CTAB 法[9]提取基因组。
2.2 DNA 条形码方法建立
从NCBI 数据库上下载风藤和山蒟的成熟酶matK基因、petA-psbJ、核糖体间隔序列等已知序列进行多序列比对,寻找差异位点附近的保守区域自行设计引物。psbA-trnH的引物直接参照2020 年版《中国药典》(四部)“9107 中药材DNA 条形码分子鉴定法指导原则”。引物合成委托上海生物工程公司进行。PCR 引物序列及扩增条件见表2。

表2 PCR 引物序列和扩增条件Tab.2 PCR primer sequences and amplification conditions
PCR 产物委托上海生物工程公司进行测通,测序引物为PCR 反应引物。去除测序结果两端信号弱或重叠峰区域。在MEGA11 软件上采用Clustal W 方法比对分析序列,采用K2P 法,bootstrap = 1 000 参数绘制Neighbor-Joining(NJ 进化树)。
2.3 SSR 方法建立
2.3.1 引物设计 根据NCBI 数据库里的风藤(KT223569.1)和山蒟(MZ046380.1)叶绿体全基因组上的简单重复序列差异位点设计引物进行PCR 扩增和毛细管电泳检测,再根据有无扩增产物长度差异筛选出四对引物,见表3。
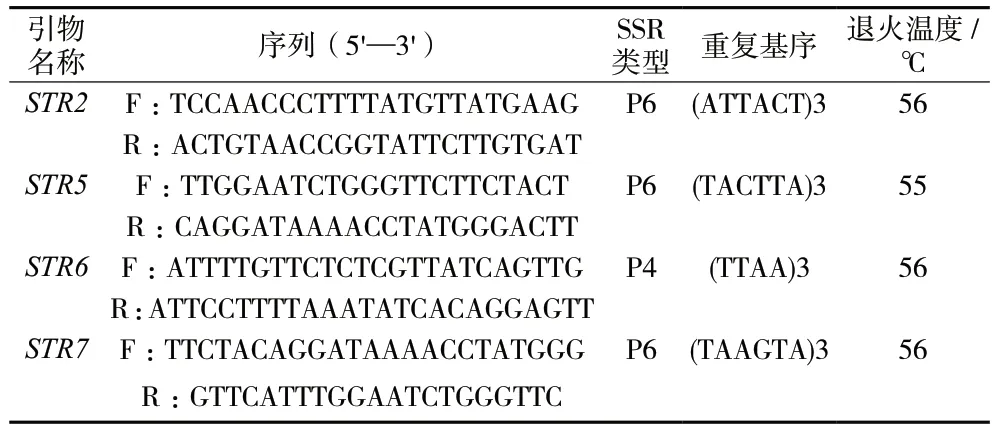
表3 4 对SSR 引物相关信息Tab.3 The information of 4 pairs of SSR primers
2.3.2 PCR 扩增条件及分析方法 PCR 扩增反应体系:DNA 模板1.0 μL(20 ~ 50 ng/μL)、5'端带FAM 的上游引物0.5 μL(10 μmol/L)、下游引物0.5 μL(10 μmol/L)均由上海生物工程有限公司合成、dNTP(mix)0.5μL(10 μmol/L)、10×Taq buffer(with MgCl2)2.5 μL、Taq 酶0.2μL(5 U/μL)、ddH2O 加至25μL。
PCR 扩增程序:预变性95℃ 5 min;变性94℃30 s,55℃退火30 s,延伸72℃30 s,循环34 次;72℃延伸 10 min。
PCR 产物进行毛细管电泳及荧光检测(上海生物工程有限公司)。
数据分析:使用Genemapper软件分析SSR多态性。
3 结果
3.1 PCR 扩增与测序
petA-psbJ片段的信息位点最多,占比5.28%,psbA-trnH片段的信息位点和序列间差异最小几乎为0。所有片段的平均序列差异均小于3%,见表4。
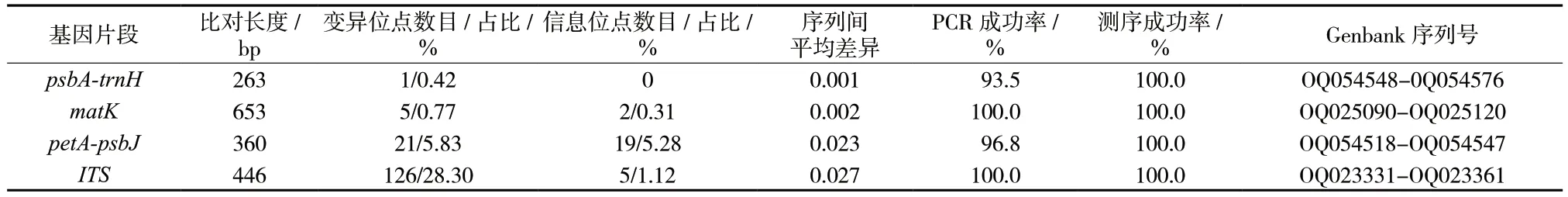
表4 DNA 条形码的特征比较Tab.4 Feature comparison of DNA barcodes
3.2 系统进化树
从系统进化树可知,psbA-trnH片段中所有样品都与风藤和山蒟聚为一支,因序列高度一致而无法对风藤和山蒟进行区分,见图1。matK和petA-psbJ片段都将样品F6、Y1、Y2、Y3、Y4、Y5、Y6、Y7 与风藤聚为一支,见图2、图3。而将除F2 外的其它样品与山蒟聚为一支,自展率均大于64%。在ITS进化树中,所有样品(除F2)都能与风藤和山蒟聚为一大簇,但无法具体归类到风藤或者山蒟一支,见图4。
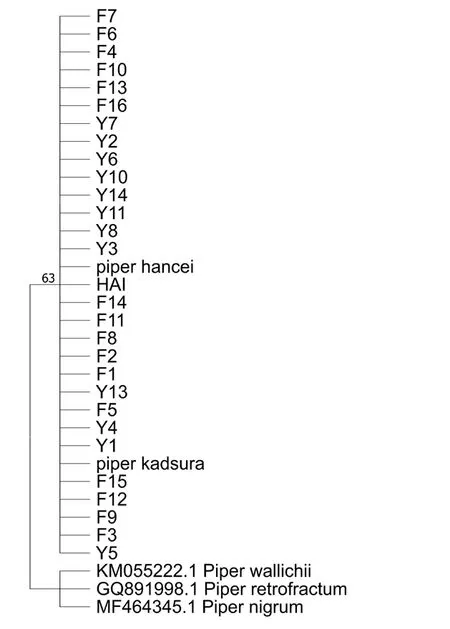
图1 基于样品和NCBI 的风藤、山蒟、石楠藤、黑胡椒,假荜拔的部分psbA-trnH 序列的NJ 进化树Fig.1 NJ trees based on part of psbA-trnH sequences of samples and Piper kadsura, Piper hancei, Piper wallichii, Piper nigrum, Piper retrofractum from NCBI

图2 基于样品和NCBI 的海风藤、山蒟、石楠藤、胡椒的部分matK 序列的NJ 进化树Fig.2 NJ trees based on part of matK sequences of samples and Piper kadsura, Piper hancei, Piper wallichii and Piper nigrum from NCBI

图3 基于样品和NCBI 的风藤、山蒟的部分petA-psbJ 间隔序列的NJ 进化树Fig.3 NJ trees based on petA-psbJ sequences of samples and Piper kadsura, Piper hancei from NCBI

图4 基于样品和NCBI 的风藤、山蒟、石楠藤、胡椒的ITS 间隔序列的NJ 进化树Fig.4 NJ trees based on ITS sequences of samples and Piper kadsura, Piper hancei, Piper wallichii and Piper nigrum from NCBI
3.3 基于叶绿体基因组的SSR 分子标记技术
根据四对引物扩增片段的大小,31 批样品可分为三大类:F6、Y1、Y2、Y3、Y4、Y5、Y6、Y7 这8 个样品扩增产物大小一致,为第一类;F2 样品的扩增片段与其它样品差异均较大,为第二类;其余样品扩增片段大小一致,为第三类。通过对风藤叶绿体基因组(NC_027941)和山蒟叶绿体基因组(MZ046380)进行四对引物的扩增模拟分析,结果显示第一类样品与风藤一致,第三类样品与山蒟一致,见表5。
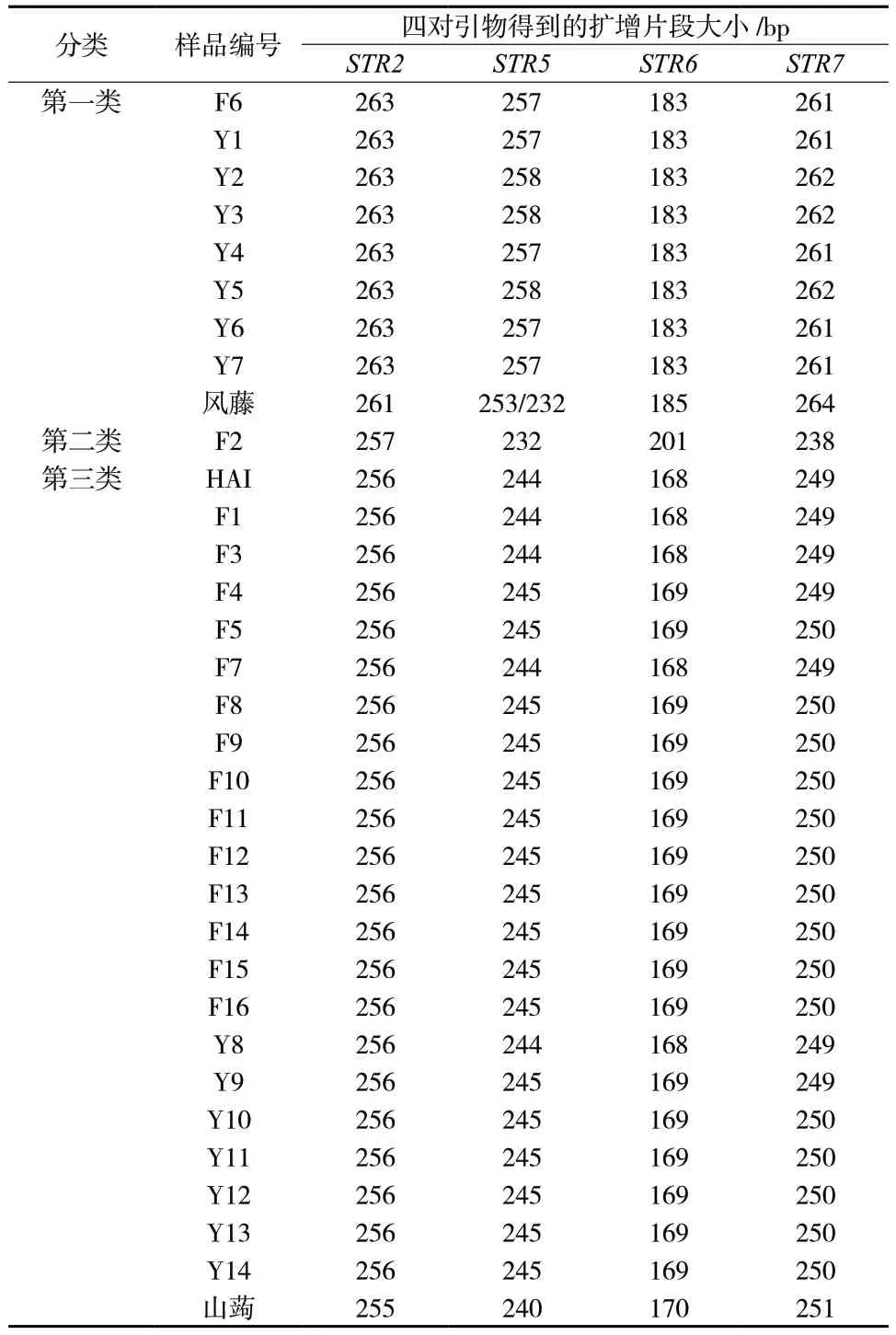
表5 31 批样品的SSR 扩增片段长度及NCBI 数据库里风藤与山蒟叶绿体基因组的SSR 扩增片段长度Tab.5 SSR amplification results of 31 samples and chloroplast genome of Piper kadsura and Piper hancei from NCBI
4 讨论
SSR 也称微卫星DNA,其串联重复的核心序列为1 ~ 6 bp,每个微卫星DNA 的核心序列结构相同,其高度多态性主要来源于串联数目的不同。DNA 条形码技术是利用基因组中一段或几段公认的、标准的、有足够变异的、易扩增且相对较短的DNA 片段,以自身在物种种内的特异性和种间的多样性而创建的分子鉴定技术,可以对物种实现准确、快速、简便的鉴定,不受发育阶段、供试部位、环境条件等的限制,弥补了传统鉴定方法在这方面的不足[10]。在被子植物中,ITS区既有核苷酸序列的高度上基于30 份样品和NCBI 的风藤、山蒟的部分petA-psbJ间隔序列的NJ 进化树度变异性,又有长度上的保守性,已成为最广泛的应用于被子植物系统发育和进化研究的核基因标记之一[11]。叶绿体基因组上编码区的核酸替代速率相对较低,为植物深层系统进化研究提供了合适的途经[12]。matK基因是叶绿体基因组的蛋白编码区中进化最快的基因之一[13]。而叶绿体基因间区如psbA-trnH等不参与转录翻译过程,因而在功能上的限制较少,变异较自由,比编码基因能够提供更多的系统发育信息[12]。
本研究选择叶绿体基因组中进化较快的编码基因matK和非编码基因间区psbA-trnH、petA-psbJ,以及核基因组上变异最多的核糖体间隔序列ITS作为DNA 条形码。四条DNA 条形码中petA-psbJ具有最大的信息位点占比5.28%和第二大的平均序列差异0.023,ITS 的信息位点占比仅在1%左右,且平均序列差异0.027 主要来源于F2 与其它序列的较大差异,psbA-trnH和matK的信息位点占比和平均序列差异都接近于零。由此推测风藤和山蒟的基因组序列差异较小,考虑到它们同样高度相似的外形特征和药理活性,建议将两者同时作为中药材海风藤的种质资源。
通过构建系统进化树,条形码matK和petA-psbJ都将舟山地区的植物原株与风藤聚为一支,其它样品(除F2)与山蒟聚为一支,且分类结果与SSR 分子标记结果一致。按照该结果,市场上的大部分药材(包括海风藤对照药材)都应该属于山蒟,而不是《中国药典》中规定的风藤。ITS条形码也能将舟山地区的植物原株和海风藤对照药材与其它样品区分开来,但所有样品均未与风藤或山蒟单独聚为一支,只是与风藤和山蒟共同聚为一大簇。显示ITS对风藤和山蒟的鉴别能力不如matK和petA-psbJ。F2 与其它样品的DNA 条形码和SSR 扩增序列均有较大差异,在matK和ITS进化树中与石楠藤Piper wallichii距离较近,显示其可能为风藤和山蒟的近缘物种石楠藤。
5 结论
本研究通过DNA 条形码和SSR 分子标记技术对23 批海风藤浙海风藤药材及8 批风藤山蒟植物原株进行分子鉴定,结果显示除样品F2 外,其余样品都能与风藤或山蒟聚为一支。风藤和山蒟的psbA-trnH序列高度一致而无法对两者进行鉴别,核糖体间隔序列ITS虽然也有一定的序列差异,但是较难鉴别风藤和山蒟,matK和petA-psbJ基因片段的差异可以用来鉴别风藤和山蒟,且鉴别结果与SSR 分子标记结果一致。该方法为海风藤的种质资源鉴定提供了一定的分子依据。
猜你喜欢
河南农业(2023年2期)2023-03-03 04:45:56
今日农业(2022年2期)2022-11-16 12:29:47
天津市教科院学报(2021年5期)2021-11-10 07:32:40
少年文艺·开心阅读作文(2021年8期)2021-09-05 02:57:46
生物学通报(2021年9期)2021-07-01 03:24:44
今日农业(2021年6期)2021-06-09 08:05:20
特种经济动植物(2021年4期)2021-04-19 07:02:18
小学科学(学生版)(2019年5期)2019-05-21 01:00:22
少儿美术(快乐历史地理)(2019年11期)2019-04-20 12:33:20
小学生导刊(2017年13期)2017-06-15 20:29:38
