
基于GBS技术开展柚类资源群体遗传评价并发掘酸含量相关基因
2023-05-15 08:52:14江东王旭李仁静赵晓东戴祥生柳正葳
中国农业科学 2023年8期
江东,王旭,李仁静, 赵晓东, 戴祥生,柳正葳
基于GBS技术开展柚类资源群体遗传评价并发掘酸含量相关基因
江东1,王旭1,李仁静1, 赵晓东2, 戴祥生2,柳正葳3
1西南大学柑橘研究所,重庆 400712;2井冈山农业科技园管理委员会,江西吉安 343016;3井冈山大学,江西吉安 343016
【目的】弄清我国柚类种质资源的起源与演化、群体遗传结构和多样性水平,高效利用柚类自然资源群体发掘与果实重要品质性状相关的基因,为柚类种质资源群体的遗传结构研究、起源演化和柚类种质创新提供重要的理论及实践依据。【方法】利用国家果树种质重庆柑橘资源圃中保存的具有广泛遗传多样性和不同地理来源的282份柚类资源作为材料,采用R I限制性内切酶消化基因组DNA后构建GBS文库,然后进行Illumina HiSeq PE150二代测序获得短读序列,通过BWA软件将序列映射到柚参考基因组上,利用SAMTOOLS软件鉴定SNP位点。依据SNP的基因分型结果,进行主成分分析和群体结构分析,并采用邻接法构建系统演化树,分析亚群的遗传多样性水平。选择23个低酸种质和32个高酸种质构成两个群体进行Fst、XP-CLR分析,同时利用282个柚类种质的基因型数据与果实可滴定酸含量的表型数据进行GWAS全基因组关联分析。【结果】利用GBS简化基因组测序技术对282份柚类种质进行测序,获得201.66 Gb的原始测序数据,每个样本的平均测序数据量为0.72 Gb,经过测序深度为5×次要等位基因频率>0.01、Miss0.2的筛选条件过滤,最终获得121 726个高质量的SNP位点。主成分分析、群体结构分析和聚类分析均表明282份柚类资源可被分为6个亚群,其中柚与‘清见’杂交群体、葡萄柚等橘柚杂种群体可明显区别于其他真正柚类种质。不同地理来源和特定类型的柚类种质在遗传水平上存在明显差异,来源于泰国、越南等东南亚国家的柚类资源形成了较为独特的类群,与国内的沙田柚类、文旦柚类、垫江柚类等群体能够明显区分开。在中国南方的大部分柚类种质中有来自于越南柚的基因渗入,表明越南是柚的原始起源中心。另外,一些来源不清的柚类种质也通过GBS技术得以准确鉴定。Fst、XP-CLR选择性清除分析发现在7号染色体上的强烈选择信号区域中包含有丙酮酸脱氢酶复合物二氢硫辛酰赖氨酸残基乙酰转移酶(PDH-E2)和铝激活苹果酸转运蛋白(ALMT9),涉及柠檬酸的合成和运输。GWAS全基因组关联分析在2号染色体上鉴定了一个与果实酸度高度关联的区域。【结论】GBS技术为研究柚的系统发育和演化提供了一种可靠和高效的方法。本研究表明人工杂交育种、长期人工选择和驯化、地理隔离是形成不同类型柚类种质的驱动力,同时也是导致柚类种质遗传分化和多样性增加的重要原因。通过Fst、XP-CLR选择性清除和全基因组关联分析鉴定了多个与柚类果实柠檬酸含量相关的候选基因,为柚类的遗传改良和育种提供了重要的基因资源。
GBS;柚系统演化;果实酸度调控基因;GWAS
0 引言
【研究意义】柚是柑橘属的4个基本种之一[1-2],其原生起源中心在南亚和东南亚国家的马来西亚、印尼、泰国、越南等地,中国是世界柚类资源的次生演化中心。柚在传播引入到我国后,与我国原有的宽皮橘、香橙、宜昌橙等资源进行天然杂交而逐渐演化形成了甜橙[1-3]、橘柚、酸橙[4-5]、香圆[6]等不同类型的柑橘种质,尤其对宽皮柑橘中杂柑的起源演化产生了重要影响[1,7-8]。由于柚种子为单胚,其实生后代的变异十分丰富,不同地区在引种过程中常通过实生选种获得新的类型,在长期的引种栽培驯化过程中,不同地区的柚类种质由于地理隔离而保留了明显的遗传印迹,逐渐形成了独具特色的地方品种。我国有丰富多样的柚类种质资源,这为研究柚的起源和演化提供了重要材料;但在以往的引种过程中,常用种子进行实生繁殖,也造成了柚类品种同物异名、同名异物的现象十分突出,对资源的保存、鉴定带来一定难度,对这些种质进行遗传多样性分析,有助于了解柚类种质的遗传背景和差异,从而为柚类资源的高效保存、基因资源挖掘提供科学的指导。长期以来,园艺学上对柚类品种的认识和划分多依据形态学特征,由于表型数据易受外界环境因素的影响,不利于柚类种质资源的精准鉴定与评价。同时柚类树体高大,田间资源保存占用土地面积较多,为加强柚类核心种质的构建,有必要弄清柚类种质群体的遗传背景和其中的特异资源,这对提高柚类种质的高效保存与利用都具有重要意义。在柚类果实中,柠檬酸含量是影响果实品质的重要因子,在柚类种质资源中存在着低酸和高酸两类种质,这为利用自然资源群体开展基因型和表型的关联分析,进而发掘与果实酸代谢相关的基因奠定了基础。本研究利用GBS简化基因组测序技术开展柚类种质的基因型鉴定和遗传多样性分析,将对柚类种质的收集保存、精准鉴定、起源演化、基因挖掘以及资源的高效利用提供重要信息。【前人研究进展】前期国内对柚类种质资源常采用分子标记技术开展资源多样性的研究,获得了一些初步结果。比如Yu等[9]利用31个SSR分子标记对274份国内柚类种质进行了遗传多样性分析,发现国内柚类种质可分为3个亚群,这3个亚群与地理起源分布存在一定相关性。刘勇等[10]利用SSR分子标记对122份我国柚类资源及近缘种进行了遗传多样性分析,发现柚类资源主要由沙田柚品种群、文旦柚品种群和杂种柚品种群组成。GBS技术是近几年来在第二代深度测序基础上发展起来的简化基因组测序技术,通过DNA酶切后加上代表样品的分组标签,实现了多样本的高通量平行测序,不仅降低了测序成本,而且能够对大量样本进行同时全基因组分型,为开展资源的遗传研究提供了重要手段和方法[11-12],利用GBS技术深入了解资源的遗传背景和群体遗传结构、多样化水平,对指导资源的收集、保存、利用都具有重要意义[13]。目前利用GBS测序技术进行SNP发掘和基因分型已经广泛应用在油橄榄[12]、柑橘[13]、枣[14]、油菜[15]、小麦[16-17]、菜豆[18]等作物的遗传多样性和群体结构研究中。若联合精准的表型鉴定和全基因组关联分析(GWAS),该技术还能够加快对园艺作物重要性状基因的挖掘[19-21]。另外,GBS技术还在全基因组选择[22]、遗传图谱构建[23-25]、遗传图谱加密[26]、基因组的辅助组装[27]、品种资源鉴定[28-29]等研究领域发挥着重要作用。在柑橘类果实中,前期发现液泡P-ATP酶citPH1和citPH5对果实酸度有重要影响[30],在柚类中是否还有其他的调控果实酸度的基因存在,值得深入研究。【本研究切入点】作物的遗传多样性是品种改良的基础,我国有丰富的柚类种质资源,对这些种质资源的精准鉴定和深入评价将促进资源的深度利用,对提高柚类品种的育种效率具有重要意义。目前利用GBS技术开展柚类资源的遗传多样性研究还未见报道。【拟解决的关键问题】利用GBS技术对国家柑橘种质资源圃中保存的282份柚类资源进行简化基因组测序,获得不同柚类品种的基因型数据,利用SNP基因型数据开展柚类群体和不同亚群的多样性分析,为阐明我国柚类种质的来源、遗传多样性水平,进而指导柚类核心种质的构建和柚类品种资源的保护提供重要的理论参考和依据。同时利用基因型和表型关联分析发掘出与果实酸含量相关的基因,为柚类的低酸品种创新提供育种辅助选择分子标记。
1 材料与方法
1.1 试验材料
为使本研究的柚类品种资源具有更广泛的遗传多样性和代表性,采用了282份柚类资源材料进行研究,这些种质是在不同时期,从越南、泰国以及国内不同地区的资源调查活动中收集,并保存在国家柑橘种质资源圃(重庆)中;另外,材料中还包括了柚的天然杂种和人工杂交材料。选择的282份柚类种质中包括真正的柚类种质194份、‘清见’与柚的人工杂交群体材料48份、与柚类亲缘关系较近的40份葡萄柚及类似杂种。另外,还有来源于越南的柚类种质14份,来源于泰国的柚类种质10份,研究材料名单见附表1。
1.2 试验方法
1.2.1 文库构建及GBS测序 田间采集柚类资源无病虫斑的健康春梢嫩叶,每份材料收集0.3—0.5 g,立刻保存于离心管置于液氮中带回实验室,将冻干的叶样组织用不锈钢珠研磨成粉末。DNA提取采用Magen Plant DNA Mini Kit试剂盒,提取的DNA样品送诺禾致源公司进行后期的GBS建库测序。DNA样品采用R I酶切后加接头标签,扩增后回收350—400 bp长度的DNA条带,这些条带经过纯化后上Illumina HiSeq测序平台上进行双末端150 bp测序。为验证GBS测序的准确性和可靠性,在测序样本中加入生物学重复,同时一并建库进行分析。
1.2.2 单核苷酸多态性(SNP)鉴定 依据建库样品和对应条形码接头序列聚合相同的样本,利用fastqc软件进行质控,去除低质量序列,剩下的序列采用BWA[31]程序将其匹配对齐到柚参考基因组上(ftp://ftp.bioinfo.wsu.edu/www.citrusgenomedb.org/Citrus_maxima/),采用参数为:mem-t4-k32-M。SNP调用采用Samtools软件进行,最终获得包含所有SNP变异及位点的VCFs文件。为获得高质量的SNP进行后续分析,采用vcftools软件设置筛选条件[32],以测序深度大于5、缺失率小于0.2、最小等位基因频率大于0.01为筛选条件,获得的SNP位点用于进一步的分析。
1.2.3 群体进化树分析 为估测282份柚类资源的系统演化,根据SNP基因型采用TreeBest软件计算距离矩阵,利用获得的遗传矩阵采用邻接法构建系统进化树,设置自展值初始值为1 000。
1.2.4 主成分分析 通过GCTA软件计算特征向量以及特征值,并利用R软件绘制PCA分布图。
1.2.5 群体遗传结构分析 利用PLINK(https://zzz. bwh.harvard.edu/plink/)[33]将vcf文件转换成bed输入文件,然后利用admixture(http://dalexander.github.io/ admixture/)软件交叉验证选择最优的K值,设置K值初始值为3—10,根据ΔK划分群体,最终选择K=3和K=6分别进行作图。
1.2.6 群体遗传多样性分析 利用Plink对选择的SNP等位基因数、基因型频率、基因多样性(GD)或预期杂合度(He)、次等位基因频率(MAF)等进行计算。为确定基因是否存在中性进化,采用vcftools软件计算6个亚群的核酸多态性值和Tajima’s D值,使用“--site-pi”选项估计每个SNP位点的核苷酸多样性π值,取平均值作为群体的π值。设置窗口长度为1 000计算Tajima’s D值,利用Plink计算群体间的分化系数Fst。
1.2.7 利用自然选择和GWAS进行果实酸含量候选基因的定位 为开展与果实酸含量相关的基因挖掘,根据282份柚类种质在重庆北碚进行多年的果实酸含量的分析数据,选择低酸(23份,可滴定酸含量<0.50%)和高酸(32份,可滴定酸含量>1.00%)种质构成两类亚群进行Fst、XP-CLR计算。计算群体间的Fst值是目前最基础、也是最常用的检测自然选择的方法,Fst可以对分层群体中不同亚群间基因相关性进行度量,Fst的取值范围为0—1,数值越大,表明群体间分化程度越大。利用plink软件逐位点计算Fst,获得Fst值后,利用R软件包qqman完成曼哈顿图的可视化。利用柑橘基因组数据库(https://www.citrusgenomedb.org/)对筛选获得的显著性较高的SNP位点进行基因注释。利用XP-CLR方法对Fst鉴定出的7号染色体进行选择清除分析,利用plink软件从高酸和低酸两个群体中提取基因型和基因位点为输入文件,选择扫描窗口为50 kb,步长为10 kb进行XP-CLR分析,计算出XP-CLR正则化值后利用R软件包进行可视化。对筛选获得的XP-CLR最大值对应的位点利用柑橘基因组数据库进行注释。
对282份柑橘种质的可滴定酸表型数据与基因型数据采用混合线性模型MLM进行GWAS全基因组关联分析,MLM公式如下:
y=Xα+Zβ+Wμ+e
通过关联的显著度筛选出潜在的候选SNP,以-log10(P)>5为设定阈值,筛选出候选的SNP位点后,再对关联位点两端的20—30 kb连锁不平衡区段筛选出候选基因。
1.2.8 芽变材料和同物异名材料的检测 利用king软件(http://people.virginia.edu/~wc9c/KING/Download. htm)检查不同种质直接的亲缘关系,从而推导出芽变材料和同一家族姊妹系材料,进而清楚地掌握种质材料的遗传背景和亲缘关系,该结果除了用于GWAS中亲缘关系的预测[34],还可有效预测芽变材料和同物异名的材料。
2 结果
2.1 测序质量
282份柚类种质经过DNA质量检测、酶切建库和GBS测序共产生201.66 Gb的测序数据,平均每个样本为0.72 Gb,去除低质量的测序数据后,保留下的高质量序列数据量为201.65 Gb,样本产生的高质量测序数据量为317.59—1 237.88 Mb,平均每个样本产生的高质量测序数据为715.05 Mb。本研究获得的GBS测序质量较高,Q20≥93.31%,Q30≥83.39%且GC分布正常。将高质量的测序数据通过BWA软件比对到柚参考基因组,该参考基因组大小为345.78 Mb,群体样本与柚参考基因组的平均比对率为97.67%,变异范围为94.52%—99.03%,说明本研究中的282份柚种质的测序质量较高,且不同柚种质资源间的遗传相似度较高。样本的平均测序深度为8.31×,序列覆盖度为17.74%—27.56%,至少有一个碱基的平均覆盖度为22.93%,至少有4个碱基的覆盖度为8.49%—17.94%,平均覆盖度为13.44%。其中彭县柚(131)的比对率最高,‘清见’杂交柚(282)的比对率最低。经samtools软件检测,在282个柚类种质中获得了3 007 528个SNP变异位点,经过测序深度为5、缺失率<0.2、次要等位基因频率>0.01的条件筛选后,最终获得121 726个高质量的SNP位点。
检测发现SNP在染色体上呈不均匀分布,在着丝粒附件的SNP数量相对较少。每条染色体上的SNP平均数量为12 841,最少为8号染色体,其上的SNP数量为8 581;2号染色体的SNP数量最多,为21 237;7号染色体的SNP数量为9 168。对获得的121 726个SNP位点进行注释,位于基因上游的SNP有7 428个,基因下游的SNP有6 746个,在外显子中导致密码子提前终止的SNP有197个,终止子丢失的SNP有45个,同义突变的SNP有12 046个,非同义突变的SNP有13 446个;而存在于内含子中的SNP有25 443个,基因间区的SNP有48 794个。单碱基替换产生的转换更多,其中转换的SNP有79 124个,颠换的SNP有42 602个,转换/颠换的比值为1.857。观察到A→G(23 665)和T→C(23 814)的转换较G→A(15 733)和C→T(15 912)的转换多。
2.2 主成分分析
利用PCA分析法对282份柚类种质进行主成分分析,其中前3个主成分能解释大部分变异。根据第一主成分和第二主成分可将282份柚类资源分为3个亚群,分别为真柚群体、葡萄柚群体、‘清见’与柚杂交群体。而根据第一主成分和第三主成分,又可以将真柚亚群细分为4个亚群,分别是沙田柚亚群、越南柚和泰国柚亚群、文旦柚亚群、垫江柚亚群。综合前3个主成分,可将282份资源分为6个亚群,分别是葡萄柚亚群、‘清见’与柚杂交群体、沙田柚亚群、文旦柚亚群、越南泰国柚亚群、垫江柚亚群(图1)。

左图依据第1和第2主成分构图,右图依据第1和第3主成分构图。红色圆圈代表‘清见’与柚杂交样本,黑色圆圈代表真柚类样本,蓝色和其他颜色圆圈代表葡萄柚样本
2.3 群体遗传结构分析
为评价282份柚类的群体结构,使用ΔK来划分亚群,当K=3时观察到最大的ΔK,表明282份柚类种质可分为3组,分别对应是葡萄柚亚群、‘清见’与柚杂种亚群和真柚亚群(图2,K=3),结果与PCA的结果吻合。若要细分真柚亚群,可设置K=6时,将282份柚类种质分为6个群体(图2,K=6)。沙田柚类群(红色)为单独一个大类,其中包括真龙柚、合江柚、岭南沙田柚、长寿沙田柚等类型以及沙田柚的杂种如脆柚、沙暹柚、沙田柚×甜橙、正形沙田柚、杭晚蜜柚实生2号、卫寺蜜柚、梁沙柚、舒化柚、早熟柚、金沙柚等,该类群果实形态多为梨形,具有明显的沙田柚的果形特征;第二类为垫江柚类群,包括垫江白柚、垫江红心柚、垫江周家白心柚、垫江曾家白柚、垫江黄沙白心柚、龙安柚以及灌香柚、北碚柚、蓬溪柚、贡水红柚、江津红心柚、金堂绿柚等,果形多为长椭圆形或卵圆形;第三类为以琯溪蜜柚为代表的文旦柚类型,包括玉环文旦、三红蜜柚、福建文旦、红肉琯溪、红绵蜜柚、黄肉蜜柚、通贤柚以及文旦柚杂种,包括左氏柚、尖顶梁平柚2号、四季抛、坪山柚等,果形多为锥状卵圆形;第四类是‘清见’与柚的杂种群体,其中包括鸡尾葡萄柚等材料,这类材料中除了有柚类的基因外,还含有其他柑橘种类的基因渗入,比如橙类的基因,果形比一般真正柚类果实偏小,果肉颜色多为橙色,果肉细软多汁;第五类包括来源于东南亚国家越南和泰国的柚类品种,越南柚类包括光皮柚、裴红柚、平阳柚、囊内柚、越南小柚、越南小甜柚、绿柚;泰国柚包括泰国柚、暹罗低酸柚、高浦柚、永安柚、泰国柚kitik、强德勒柚、彭县暹罗柚,以及与东南亚国家邻近地域的品种,如勐仑早柚、晚白柚等,其中国内的一些柚类品种也包括其中,表明这些品种中含有越南、泰国柚等外源基因的渗入,可能最早是通过品种引进再与国内品种杂交后驯化而成。这些品种如川渝地区的梁平柚、江北无核柚、纳溪樱桃柚、梅塆柚、蒲莲柚、金堂绿柚、‘清见’ב强德勒’,湖南安江地区的安江香柚、安江石榴柚、安农1号香柚、大庸菊花心、安农无核蜜柚、白玉霜、锅魁柚,浙江、江西等地的永嘉早香柚、金兰柚、水晶文旦、盘谷文旦以及一些杂柚品种如8088柚、奇龙嘉、脐柚等。这个类群中的基因来源复杂,但主要以越南柚中的基因为主要遗传构成。这个类群多数柚类品种果形为高扁圆形,成熟期普遍偏晚,表明起源于南亚热带地区的柚类品种在中亚热带气候条件下,成熟期有推迟趋势;第六类包括葡萄柚类型品种,主要有葡萄柚、胡柚、温岭高橙、建阳橘柚、‘清见’与柚的杂种、‘日本4’×夏橙、奥罗勃朗柯、世界蜜柚等,在这个类群中,比如像建阳橘柚具有橘类的基因渗入,‘日本4’×夏橙具有更多橙类基因的渗入。
通过对群体结构的进一步分析,发现相同地理来源的柚类品种通常聚在一起,比如来自垫江及其周边的柚类品种多聚在一起;来自越南和泰国的柚类品种通常聚集在越南和泰国柚类群下,不同地理来源的品种存在较明显的遗传差异。
2.4 群体进化树分析
采用邻接法构建了群体进化树,经过自展值达1 000次的计算获得。群体进化树结果表明,282份柚类资源种质聚为6个大类。其中第一个类群为川渝地区的地方种质,包括垫江柚、垫江柚杂种、北碚柚、莲蒲柚、梅湾柚等;第二类为沙田柚及其杂种类型,另外与沙田柚地理分布位置较近的越南柚、勐仑早柚、绿肉柚、暹罗蜜柚也与其聚在一起。第三类由琯溪蜜柚等文旦柚类型组成,另外还包括来自于湖南的部分柚类品种,如大庸菊花芯柚等。第四类由泰国柚类,如暹罗低酸柚、强德勒柚、泰国柚及一些与真正柚类遗传距离较远的种质构成,包括水晶文旦柚、枳雀等柚类杂种。可见在真正的柚类种质资源中,大致可以划分为泰国柚类群、文旦柚类群、越南柚与沙田柚类群、垫江柚类群。第五类由‘清见’与柚的杂交群体构成。第六类大部分由葡萄柚及其类似杂种组成。另外,从群体的聚类图中看,水晶文旦、枳雀、285号柚品种在遗传水平上存在明显的异质性,这些种质应该含有更多外源基因的渗入,可以作为核心种质加以重点保存。从群体分化的结果来看,地理隔离、种间杂交、引种驯化是引起柚类种质遗传分化的重要驱动力。
2.5 群体的遗传多样性
对282份柚类种质进行群体的遗传多样性评估,基因多样性GD值(即期望杂合度He)在282份柚类种质中变化范围从0.02—0.5,平均值为0.2103;PIC值变化范围从0.02—0.38,平均值为0.178;MAF值的变化范围在0.01—0.50,平均值为0.139。利用Vcftools对6个不同亚群的p值和Tajima’s D值进行计算(表2)。
从表2可见,6个亚群的p值变化范围处于0.01—0.51,葡萄柚亚群的平均核酸多态性p 值最大,其次是‘清见’杂交柚类群,这主要归因于两个群体均来源于柚与其他柑橘的种间杂交,造成基因组遗传分化和多样性水平的增加,因而后代个体在遗传组成上与真正柚类种质的差异较大。沙田柚亚群的平均p值最低,其次是文旦柚亚群,表明在沙田柚群体和文旦柚群体中可能经历了较大的人工选择压力或者人为繁殖导致基因组核酸多态性水平的降低。基因组的Tajiama’s D值往往与外源基因的渗入呈正相关,本研究发现‘清见’杂交柚群体的Tajima’s D值最大,平均值为1.12,表明杂交群体中个体间的异质性较大,群体的多样性水平较高。葡萄柚群体Tajima’s D值为0.92,文旦柚群体的Tajima’s D平均值为0.41,沙田柚群体的Tajima’s D平均值为-0.15,表明沙田柚群体由于长期的大量引种、栽培面积的扩大和人工选择压力的作用,亚群内个体间的异质性较小,基因组上存在选择性清除效应。利用Plink计算群体间的分化系数Fst,越南、泰国柚群体与葡萄柚群体的Fst值最大为0.24,其次是垫江柚群体和‘清见’杂交柚群体的Fst值为0.22,沙田柚群体和文旦柚群体、垫江柚群体的Fst值接近,分别为0.18和0.16。垫江柚群体与越南、泰国柚群体的Fst值最小,为0.06,其次是文旦柚群体与越南、泰国柚群体的Fst值为0.08,‘清见’杂交柚群体与葡萄柚群体的Fst值为0.09。一般认为Fst大于0.2,群体间即存在明显的遗传分化,葡萄柚群体和‘清见’杂交柚群体与真正柚类亚群的Fst值均大于0.2。Fst值大小可反映亲缘关系的远近,可见国内真正柚类品种群与越南、泰国柚类群的分化远小于柚类与葡萄柚、‘清见’杂交柚的遗传分化。

图3 利用282份柚类种质的121726 SNP采用邻接法构建群体进化树

表2 6个柚类亚群的遗传多样性
2.6 利用自然选择法对可滴定酸候选基因的初步定位
果实中有机酸的含量是决定果实品质和风味的一个重要性状,柚类果实中的可滴定酸绝大部分为柠檬酸[35]。通过对282份柚类种质在重庆北碚地区多年的果实可滴定酸含量分析,利用Plink计算低酸(23)和高酸(32)两个群体的Fst值,根据最大的Fst值,鉴定了4个可滴定酸的候选基因,均位于7号染色体上。在7号染色体上与可滴定酸含量显著关联的SNP位点,位于Cg7g014530.1和Cg7g014550.1的基因间区,其靠5′端的邻近基因为Cg7g014530.1,该基因注释为丙酮酸脱氢酶复合物(PDC)的一个亚基—二氢硫辛酰乙酰转移酶(PDH-E2),该酶是柠檬酸合成中的一个重要酶,PDH-E2通过丙酮酸生成乙酰辅酶A,而乙酰辅酶A、草酰乙酸和H2O在ATP-柠檬酸合酶的催化下,在线粒体中以双向反应形成CoA和柠檬酸。由于该酶是参与乙酰辅酶A生成的重要蛋白,因此对柠檬酸的合成十分重要。对该SNP位点的基因型分析表明,基因型为CC、TC的为高酸品种,而基因型为TT的为低酸品种。Fst分化系数最大的SNP位于7号染色体16 299 216处的Cg7g014580,该基因注释为丝氨酸/苏氨酸蛋白激酶PRP4同源物。在该基因附近有Cg7g014630.1,注释为铝激活苹果酸转运蛋白4,在这个基因的下游是Cg7g014640.1,基因注释为丝氨酸/苏氨酸蛋白磷酸酶2A 65 kDa调节亚基PP2A。利用XP-CLR方法在低酸(23)和高酸(32)两个类群中进行选择性清除分析,同样鉴定出PDH-E2所在基因位置有最强的选择信号。
GWAS鉴定了与果实可滴定酸含量显著关联的一系列关联位点。其中2号染色体上1 168 947—1 170 318有最大的关联信号。对前后20 Kb区段进行基因注释,其中最强关联信号的基因注释为线粒体DGP2_ARATH DAR GTPase 2,该区段内还包括编码丝氨酸/苏氨酸蛋白磷酸酶PP2A的同源基因Cg2g000890以及F-box/kelch-重复蛋白的基因。而丝氨酸/苏氨酸蛋白磷酸酶PP2A在Fst和XP-CLR选择清除的显著区段也被发现与可滴定酸的含量显著相关。Fst、XP-CLR和GWAS结果均表明PP2A与果实中可滴定酸含量有显著的相关性。

Fst最大值对应的SNP位于7号染色体上The corresponding maximum position of Fst and XP-CLR is on thesame location of chromosome 7
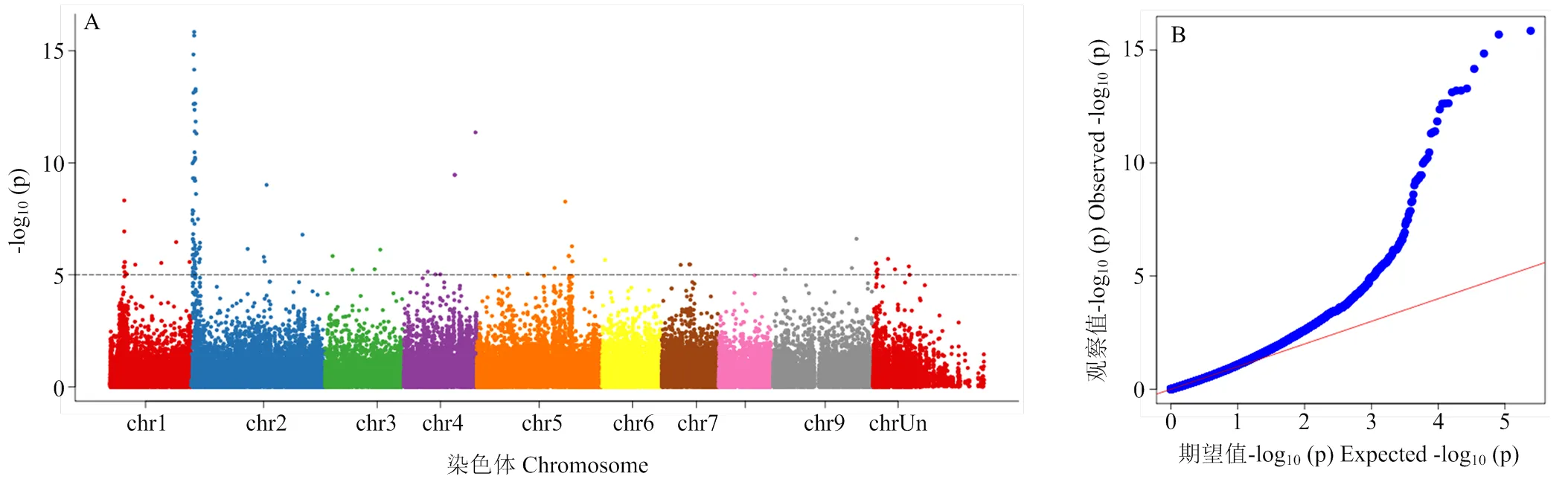
图5 利用282份柚种质采用GWAS分析鉴定柚果实可滴定酸的曼哈顿图(A)和QQ图(B)
2.6 未知亲本材料的鉴定
本研究采用King软件对柚种质个体间亲缘关系进行研究,不仅是利用自然群体进行GWAS定位的前提,也为揭示材料间的亲缘关系提供了重要线索。研究发现King能够准确对生物学重复、芽变材料、全同胞家系个体进行准确鉴定。本研究中,加入了一些生物学重复材料,比如2份晚白柚、2份胡柚、2份东风早柚、3份斐红柚都被准确鉴定出来,表明GBS的基因分型结果准确可靠。同时King能够对一些存疑品种进行区分,比如64、150、208三个品种被鉴定为生物学重复,其中64、208是龙安柚,而150是缅甸柚的概率应该很小,更大可能是龙安柚。另外,泰国柚3-Kitik与永安柚、潼南柚与琯溪蜜柚、安柚与越南小甜柚、贡水红柚和江津红心柚的相似度都较高。垫江白柚和垫江红心柚的遗传相似度也很高,其实生后代也多数聚集在一起。梁平柚与凤凰柚、虎蜜柚的亲缘系数很高,表明这些品种间有较近的亲缘关系。一些同名的品种如段氏柚,被划分到3个亚群中,明显属于同名异物,需要对这些材料进一步核实。福建文旦柚虽然被划分到1个亚类下,但两份福建文旦柚仍不完全相同,表明这两份材料尽管亲缘关系较近,可能仍然具有一定遗传差异。本研究表明利用GBS技术结合King软件来鉴定柚类品种的亲缘关系是可行的。从系统发育树和King分析的结果来看,即使柚类品种是从种子实生繁殖得来,其全同胞家系个体间的遗传组成并没有太大的变异,实生后代常与亲本材料聚在一起,暗示了柚类种质的基因纯合水平可能较高,而杂合水平可能较低。
3 讨论
3.1 不同地理区域柚类种质资源存在显著遗传差异和形态特征
不同地理区域由于气候生态环境的不同,对群体的遗传特异性塑造产生了重要影响,地理的隔离会造就独特的种群,这在许多植物和动物中都已经得到证实[36-37]。本研究表明不同地域的柚类种质具有明显不同的遗传特征,来源于泰国、越南和国内的柚类资源存在明显的遗传差异,而相同或邻近区域内的柚类种质在遗传组成上的差异往往较小,比如泰国的暹罗低酸柚、强德勒柚、泰国柚等几个品种聚在一起,而越南的光皮柚、囊内柚、绿柚等明显聚在一起。垫江白柚类群起源于重庆,由于群体后代较多,也形成了具有特色的地方品种群。沙田柚是华南地区的重要柚类品种,受人为选择压力较大或者人为的广泛繁殖,因此,该品种类群个体中保持了明显的沙田柚特征。综合来看,柚类品种存在明显的地理分布特征,柚起源于东南亚地区,在引入到国内后,为适应当地的气候和人为选择压力,演化出了新的类型,如沙田柚、文旦柚和垫江白柚等类型。
不同类型的柚类亚群具有独特的形态特征和地理起源,如沙田柚亚群起源于华南地区,树势较强,树形较直立,果形为梨形,果颈明显,果肉白色,果实酸度较低,成熟往往较晚,被引入到不同地区,形成不同的品系,如长寿沙田柚、垫江沙田柚、合江柚、真龙柚等。而文旦柚类的典型代表是琯溪蜜柚,起源于东南沿海的福建等地,树势偏弱,树形扁圆形,果皮光滑,果形尖卵圆形或扁圆形,中心柱多空虚,果实酸度较高,成熟较早。而垫江柚亚群的种质,树势强,树形较圆头或开张形,果实椭圆形,中心柱多空虚,成熟较早。这3个类群具有明显的地域特征和遗传构成。这与国内Yu等[9]、刘勇等[10]利用SSR分子标记和何天富[38]利用形态学特征对国内柚类种质进行多样性分析的结果相吻合。利用GBS的群体遗传分析结果,提出了不同地理区域的代表性核心种质,其中华南地区的代表柚类种质是沙田柚,东南沿海的代表柚类种质是琯溪蜜柚,而西南地区代表柚类种质是垫江柚。
3.2 越南柚在我国柚类起源演化中产生重要影响
地理隔离虽然塑造了品种亚群,但也存在亚群间的基因交流。本研究发现沙田柚、文旦柚、垫江白柚亚群间通过基因渐渗形成新的种质。从柚类起源演化的角度来看,来源于起源中心的柚类品种,更广泛地将其基因渗入到周边的柚类品种中。本研究中纳入了起源中心的越南和泰国的柚类资源,从群体结构分析来看,越南柚所提供的基因渗入在真正柚类群体中的作用非常明显,这可以从垫江柚群体与越南柚群体的Fst值最小为0.06,文旦柚群体与越南柚群体的Fst值为0.08得以证实,因此,越南的柚类资源对我国柚类的起源演化具有重要影响。由于越南柚的基因更多渗入我国的柚类种质中,并与沙田柚、文旦柚和垫江柚存在基因交流,由此推测,越南是柚的原初起源中心,而次生中心所演化形成的独特柚类种质,如沙田柚、文旦柚、垫江柚的基因也渗入到国内多数柚品种中,但不如越南柚的贡献更大。在沙田柚系中沙暹柚、正形沙田柚、杭晚蜜柚实生、梁砂柚等资源中都有越南柚基因的渗入,贡水红柚、江津红心柚、金堂绿柚等资源中也具有较大比例的越南柚基因的贡献;而文旦柚系中,除了坪山柚、四季抛、左氏柚、楚门文旦等几份柚外,其余品种资源中越南柚的基因贡献很少,表明地理位置的邻近对柚类种质的形成具有重要的影响和促进作用。
3.3 柚类果实中决定柠檬酸含量基因的初步定位
对柚类果实中柠檬酸含量的基因定位,以往采用混池分离法或构建杂交群体进行QTL定位方式来发掘。由于GBS能够获得高密度的SNP位点,利用性状迥异的两类自然群体通过计算Fst和XP-CLR来发掘与性状相关点的基因在动、植物中都已经证明是高效可靠的[39-40]。本研究通过对低酸和高酸柚群体的Fst和XP-CLR选择性清除分析,定位到7号染色体上与可滴定酸含量显著关联的SNP位点,其邻近的基因编码丙酮酸脱氢酶复合物(DHC)的一个亚基——二氢硫辛酰赖氨酸残基乙酰转移酶(PDH-E2),该酶主要负责乙酰-CoA的生成。由于GBS是通过对酶切位点附近的序列进行测序分型,SNP的密度相对重测序来说要低很多,在基因上有可能缺乏酶切位点而未能鉴定到SNP差异。该研究发现的SNP虽未直接定位于基因上,但这种结构变异所带来的“搭车效应”也反映出高酸品种和低酸品种群体间,在该SNP位点附近的基因有可能存在其他结构上的变异,进而对基因的功能产生影响。而该SNP位点的基因分型结果表明,基因型为CC、TC的为高酸品种,而基因型为TT的为低酸品种,证实该SNP位点可作为低酸柚品种选育的重要分子标记加以利用。柠檬酸在细胞内一般通过三羧酸循环(TCA)途径不断产生和循环。在线粒体内,丙酮酸和硫辛酰胺-E首先形成S-乙酰基二氢硫辛酰胺-E,其在PDH-E2的作用下,与(R)-硫辛酸、CoA催化形成二氢硫辛酰胺-E和乙酰-CoA,草酰乙酸、H2O和乙酰-CoA进一步在ATP-柠檬酸合酶的催化下,在线粒体中以双向反应形成CoA和柠檬酸。乙酰-CoA是柠檬酸循环的最重要的起始底物,因此,PDH-E2编码基因结构或者活性的改变,会影响其蛋白酶活性或者含量,通过调节乙酰-CoA的生成,进而影响柠檬酸的合成。另外,DHC的E1亚基还受磷酸化调控,具有无活性(磷酸化)和有活性(去磷酸化)两种形式,丙酮酸脱氢酶激酶(PDK)通过磷酸化PDH可抑制其酶活,使乙酰-COA的含量减少,进而影响柠檬酸的合成[41]。在其他果树中,如桃的低酸品种‘欧亨利’,PDK的表达量增高,减少了乙酰-COA的生产,进而影响了果实中柠檬酸的含量[42]。本研究发现7号染色体16 299 216处的丝氨酸/苏氨酸蛋白激酶PRP4,和2号染色体上的丝氨酸/苏氨酸蛋白磷酸酶PP2A都与果实中柠檬酸的高低存在一定关联,这些激酶和磷酸酶是否能对DHC的E1亚基进行磷酸化或去磷酸化,进而调控果实中酸的含量还需在后续试验中进一步研究。另外,铝激活苹果酸转运蛋白4,也可能是影响柠檬酸含量的重要候选基因。在番茄中调控苹果酸积累的主效基因编码一个铝激活苹果酸转运蛋白()[43],是导致番茄果实苹果酸含量变异的重要基因,其启动子区的GTC序列插入/缺失引起基因表达差异是影响苹果酸积累的主要因素。此外,Li等[44]还报道调控苹果酸积累的与具有高同源性,葡萄中不仅具备苹果酸转运能力,还可介导酒石酸转运[45];而在桃中,的下调,会引起苹果酸转运到液泡中的含量减少从而导致果实酸度降低[42]。本研究中,该基因在柚类中是否通过介导柠檬酸的转运,进而影响果实的酸度,也还需要继续深入研究。
4 结论
本研究利用GBS技术对282份柚类资源进行了简化基因组测序,根据系统演化树、群体遗传结构和遗传多样性分析,结合King软件能够准确地对282份柚类资源进行准确划分,证明GBS技术可高效用于资源材料的精准鉴定和亲缘关系的研究,对提高柚类资源的保存、育种效率和科学管理都具有重要价值。同时,研究证明了地理隔离、杂交育种、人工选择是柚种质资源遗传分化和多样性增加的内在驱动力,越南是柚类起源中心,在我国柚类的起源演化中扮演重要角色。中国作为柚类资源的次生演化中心,也具有独特的核心骨干种质,可根据中国柚类种质的品种特点和地域分布划分为沙田柚系、文旦柚系、垫江柚系。本研究利用Fst、XP-CLR和GWAS技术鉴定了柚类果实中与可滴定酸性状相关的重要候选PDH-E2基因和铝激活苹果酸转运蛋白4,为今后柚类品质遗传改良提供了重要的可候选基因。
[1] WU G A, TEROL J, IBANEZ V, LÓPEZ-GARCÍA A, PÉREZ- ROMÁN E, BORREDÁ C, DOMINGO C, TADEO F R, CARBONELL- CABALLERO J, ALONSO R, CURK F, DU D L, OLLITRAULT P, ROOSE M L, DOPAZO J, GMITTER F G, ROKHSAR D S, TALON M. Genomics of the origin and evolution of. Nature, 2018, 554(7692): 311-316.
[2] WANG X, XU Y T, ZHANG S Q, CAO L, HUANG Y, CHENG J F, WU G Z, TIAN S L, CHEN C L, LIU Y, YU H W, YANG X M, LAN H, WANG N, WANG L, XU J D, JIANG X L, XIE Z Z, TAN M L, LARKIN R M, CHEN L L, MA B G, RUAN Y J, DENG X X, XU Q. Genomic analyses of primitive, wild and cultivated citrus provide insights into asexual reproduction. Nature Genetics, 2017, 49(5): 765-772.
[3] XU Q, CHEN L L, RUAN X A, CHEN D J, ZHU A D, CHEN C L, BERTRAND D, JIAO W B, HAO B H, LYON M P, CHEN J J, GAO S, XING F, LAN H, CHANG J W, GE X H, LEI Y, HU Q, MIAO Y, WANG L, XIAO S X, BISWAS M K, ZENG W F, GUO F, CAO H B, YANG X M, XU X W, CHENG Y J, XU J, LIU J H, LUO O J, TANG Z H, GUO W W, KUANG H H, ZHANG H Y, ROOSE M L, NAGARAJAN N, DENG X X, RUAN Y J. The draft genome of sweet orange (). Nature Genetics, 2013, 45(1): 59-66.
[4] WU G A, SUGIMOTO C, KINJO H, AZAMA C, MITSUBE F, TALON M, GMITTER F G, ROKHSAR D S. Diversification of mandarin citrus by hybrid speciation and apomixis. Nature Communications, 2021, 12(1): 4377.
[5] BARKLEY N A, ROOSE M L, KRUEGER R R, FEDERICI C T. Assessing genetic diversity and population structure in a citrus germplasm collection utilizing simple sequence repeat markers (SSRs). Theoretical and Applied Genetics, 2006, 112(8): 1519-1531.
[6] DEMARCQ B, CAVAILLES M, LAMBERT L, SCHIPPA C, OLLITRAULT P, LURO F. Characterization of odor-active compounds of ichang lemon (Tan.) and identification of its genetic interspecific origin by DNA genotyping. Journal of Agricultural and Food Chemistry, 2021, 69(10): 3175-3188.
[7] GONZALEZ-IBEAS D, IBANEZ V, PEREZ-ROMAN E, BORREDÁ C, TEROL J, TALON M. Shaping the biology of citrus: II. Genomic determinants of domestication. The Plant Genome, 2021, 14(3): e20133.
[8] WU G A, PROCHNIK S, JENKINS J, SALSE J, HELLSTEN U, MURAT F, PERRIER X, RUIZ M, SCALABRIN S, TEROL J,. Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during citrus domestication. Nature Biotechnology, 2014, 32(7): 656-662.
[9] YU H W, YANG X M, GUO F, JIANG X L, DENG X X, XU Q. Genetic diversity and population structure of pummelo () germplasm in China. Tree Genetics & Genomes, 2017, 13(3): 58.
[10] 刘勇, 刘德春, 吴波, 孙中海. 利用SSR标记对中国柚类资源及近缘种遗传多样性研究. 农业生物技术学报, 2006, 14(1): 90-95.
LIU Y, LIU D C, WU B, SUN Z H. Genetic diversity of pummelo and their relatives based on SSR markers. Journal of Agricultural Biotechnology, 2006, 14(1): 90-95. (in Chinese)
[11] ELSHIRE R J, GLAUBITZ J C, SUN Q, POLAND J A, KAWAMOTO K, BUCKLER E S, MITCHELL S E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE, 2011, 6(5): e19379.
[12] Islam A S M F, SANDERS D, MISHRA A K, JOSHI V. Genetic diversity and population structure analysis of the USDA olive germplasm using genotyping-by-sequencing (GBS). Genes, 2021, 12(12): 2007.
[13] 王小柯, 江东, 孙珍珠. 利用GBS技术研究240份宽皮柑橘的系统演化. 中国农业科学, 2017, 50(9): 1666-1673.doi: 10.3864/j.issn. 0578-1752.2017.09.012.
WANG X K, JIANG D, SUN Z Z. Study on phylogeny of 240 mandarin accessions with genotyping-by-sequencing technology. Scientia Agricultura Sinica, 2017, 50(9): 1666-1673. doi: 10.3864/ j.issn.0578-1752.2017.09.012. (in Chinese)
[14] CHEN W, HOU L, ZHANG Z Y, PANG X M, LI Y Y. Genetic diversity, population structure, and linkage disequilibrium of a core collection ofassessed with genome-wide SNPs developed by genotyping-by-sequencing and SSR markers. Frontiers in Plant Science, 2017, 8: 575.
[15] BIRD K A, AN H, GAZAVE E, GORE M A, PIRES J C, ROBERTSON L D, LABATE J A. Population structure and phylogenetic relationships in a diverse panel ofL. Frontiers in Plant Science, 2017, 8: 321.
[16] ELTAHER S, SALLAM A, BELAMKAR V, EMARA H A, NOWER A A, SALEM K F M, POLAND J, BAENZIGER P S. Genetic diversity and population structure of F3:6Nebraska winter wheat genotypes using genotyping-by-sequencing. Frontiers in Genetics, 2018, 9: 76.
[17] ALIPOUR H, BIHAMTA M R, MOHAMMADI V, PEYGHAMBARI S A, BAI G H, ZHANG G R. Genotyping-by-sequencing (GBS) revealed molecular genetic diversity of Iranian wheat landraces and cultivars. Frontiers in Plant Science, 2017, 8: 1293.
[18] DELFINI J, MODA-CIRINO V, DOS SANTOS NETO J, RUAS P M, SANT'ANA G C, GEPTS P, GONÇALVES L S A. Population structure, genetic diversity and genomic selection signatures among a Brazilian common bean germplasm. Scientific Reports, 2021, 11(1): 2964.
[19] LUO Z N, BROCK J, DYER J M, KUTCHAN T, SCHACHTMAN D, AUGUSTIN M, GE Y F, FAHLGREN N, ABDEL-HALEEM H. Genetic diversity and population structure of aspring panel. Frontiers in Plant Science, 2019, 10: 184.
[20] HYUN D Y, SEBASTIN R, LEE G A, LEE K J, KIM S H, YOO E, LEE S, KANG M J, LEE S B, JANG I, RO N Y, CHO G T. Genome-wide SNP markers for genotypic and phenotypic differentiation of melon (L.) varieties using genotyping- by-sequencing. International Journal of Molecular Sciences, 2021, 22(13): 6722.
[21] AKRAM S, ARIF M A R, HAMEED A. A GBS-based GWAS analysis of adaptability and yield traits in bread wheat (L.). Journal of Applied Genetics, 2021, 62(1): 27-41.
[22] POLAND J, ENDELMAN J, DAWSON J, RUTKOSKI J, WU S Y, MANES Y, DREISIGACKER S, CROSSA J, SÁNCHEZ-VILLEDA H, SORRELLS M, JANNINK J L. Genomic selection in wheat breeding using genotyping-by-sequencing. The Plant Genome, 2012, 5(3): 103-113.
[23] KAUR G, PATHAK M, SINGLA D, SHARMA A, CHHUNEJA P, SARAO N K. High-density GBS-based genetic linkage map construction and QTL identification associated with yellow mosaic disease resistance in bitter gourd (L.). Frontiers in Plant Science, 2021, 12: 671620.
[24] ABED A, BADEA A, BEATTIE A, KHANAL R, TUCKER J, BELZILE F. A high-resolution consensus linkage map for barley based on GBS-derived genotypes. Genome, 2022, 65(2): 83-94.
[25] VERMA S, GUPTA S, BANDHIWAL N, KUMAR T, BHARADWAJ C, BHATIA S. High-density linkage map construction and mapping of seed trait QTLs in chickpea (L.) using Genotyping-by-Sequencing (GBS). Scientific Reports, 2015, 5(1): 17512.
[26] POLAND J A, BROWN P J, SORRELLS M E, JANNINK J L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE, 2012, 7(2): e32253.
[27] GAUR R, VERMA S, PRADHAN S, AMBREEN H, BHATIA S. A high-density SNP-based linkage map using genotyping-by-sequencing and its utilization for improved genome assembly of chickpea (L.). Functional & Integrative Genomics, 2020, 20(6): 763-773.
[28] WANG B Y, TAN H W, FANG W P, MEINHARDT L W, MISCHKE S, MATSUMOTO T, ZHANG D P. Developing single nucleotide polymorphism (SNP) markers from transcriptome sequences for identification of Longan () germplasm. Horticulture Research, 2015, 2: 14065.
[29] PEMBLETON L W, DRAYTON M C, BAIN M, BAILLIE R C, INCH C, SPANGENBERG G C, WANG J P, FORSTER J W, COGAN N O I. Targeted genotyping-by-sequencing permits cost- effective identification and discrimination of pasture grass species and cultivars. Theoretical and Applied Genetics, 2016, 129(5): 991-1005.
[30] STRAZZER P, SPELT C E, LI S J, BLIEK M, FEDERICI C T, ROOSE M L, KOES R, QUATTROCCHIO F M. Hyperacidification offruits by a vacuolar proton-pumping P-ATPase complex. Nature Communications, 2019, 10(1): 744.
[31] LI H, DURBIN R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 2009, 25(14): 1754-1760.
[32] DANECEK P, AUTON A, ABECASIS G, ALBERS C A, BANKS E, DEPRISTO M A, HANDSAKER R E, LUNTER G, MARTH G T, SHERRY S T, MCVEAN G, DURBIN R, GROUP 1 G P A. The variant call format and VCFtools. Bioinformatics, 2011, 27(15): 2156-2158.
[33] CHANG C C. Data management and summary statistics with PLINK. Methods in Molecular Biology, 2020, 2090: 49-65.
[34] MANICHAIKUL A, MYCHALECKYJ J C, RICH S S, DALY K, SALE M, CHEN W M. Robust relationship inference in genome-wide association studies. Bioinformatics, 2010, 26(22): 2867-2873.
[35] PAN T F, ALI M M, GONG J M, SHE W Q, PAN D M, GUO Z X, YU Y, CHEN F X. Fruit physiology and sugar-acid profile of 24 pomelo ((L.) osbeck) cultivars grown in subtropical region of China. Agronomy, 2021, 11(12): 2393.
[36] D'AGOSTINO N, TARANTO F, CAMPOSEO S, MANGINI G, FANELLI V, GADALETA S, MIAZZI M M, PAVAN S, DI RIENZO V, SABETTA W, LOMBARDO L, ZELASCO S, PERRI E, LOTTI C, CIANI E, MONTEMURRO C. GBS-derived SNP catalogue unveiled wide genetic variability and geographical relationships of Italian olive cultivars. Scientific Reports, 2018, 8(1): 15877.
[37] MEDINA R, WOGAN G O U, BI K, TERMIGNONI-GARCÍA F, BERNAL M H, JARAMILLO-CORREA J P, WANG I J, VÁZQUEZ- DOMÍNGUEZ E. Phenotypic and genomic diversification with isolation by environment along elevational gradients in a neotropical treefrog. Molecular Ecology, 2021, 30(16): 4062-4076.
[38] 何天富. 中国柚类栽培. 北京: 中国农业出版社, 1999.
HE T F. Cultivation of Pomelo in China. Beijing: China Agriculture Press, 1999. (in Chinese)
[39] BAAZAOUI I, MCEWAN J, ANDERSON R, BRAUNING R, MCCULLOCH A, VAN STIJN T, BEDHIAF-ROMDHANI S. GBS data identify pigmentation-specific genes of potential role in skin-photosensitization in two Tunisian sheep breeds. Animals, 2019, 10(1): 5.
[40] ZHANG M M, YANG L, SU Z C, ZHU M Z, LI W T, WU K L, DENG X M. Genome-wide scan and analysis of positive selective signatures in Dwarf Brown-egg Layers and Silky Fowl chickens. Poultry Science, 2017, 96(12): 4158-4171.
[41] NILO-POYANCO R, MORAGA C, BENEDETTO G, ORELLANA A, ALMEIDA A M. Shotgun proteomics of peach fruit reveals major metabolic pathways associated to ripening. BMC Genomics, 2021, 22(1): 17.
[42] ZHENG B B, ZHAO L, JIANG X H, CHERONO S, LIU J J, OGUTU C, NTINI C, ZHANG X J, HAN Y P. Assessment of organic acid accumulation and its related genes in peach. Food Chemistry, 2021, 334: 127567.
[43] YE J, WANG X, HU T X, ZHANG F X, WANG B, LI C X, YANG T X, LI H X, LU Y E, GIOVANNONI J J, ZHANG Y Y, YE Z B. An InDel in the promoter of Al-ACTIVATED MALATE TRANSPORTER9 selected during tomato domestication determines fruit malate contents and aluminum tolerance. The Plant Cell, 2017, 29(9): 2249-2268.
[44] LI C L, DOUGHERTY L, COLUCCIO A E, MENG D, EL-SHARKAWY I, BOREJSZA-WYSOCKA E, LIANG D, PIÑEROS M A, XU K N, CHENG L L. Apple ALMT9 requires a conserved C-terminal domain for malate transport underlying fruit acidity. Plant Physiology, 2020, 182(2): 992-1006.
[45] DE ANGELI A, BAETZ U, FRANCISCO R, ZHANG J B, CHAVES M M, REGALADO A. The vacuolar channel VvALMT9 mediates malate and tartrate accumulation in berries of. Planta, 2013, 238(2): 283-291.
Population Genomic Structure of Pomelo Germplasm and Fruit Acidity Associated Genes Identification by Genotyping-by-Sequencing Technology
JIANG Dong1, WANG Xu1, LI RenJing1, ZHAO XiaoDong2, DAI XiangSheng2, Liu ZhengWei3
1Citrus Research Institute, Southwest University, Chongqing 400712;2Jinggangshan Agricultural Science and Technology Park Management Committee, Ji’an 343016, Jiangxi;3Jianggangshan University, Ji’an 343016, Jiangxi
【Objective】To reveal the phylogeny, population genetic structure and diversity level of pomelo ((L.) Osbeck) germplasm, and to efficiently utilize them to explore genes related to important fruit quality traits, this research provided an insight into the population genetic structure and phylogeny of pomelo germplasm and facilitated the pomelo varieties innovation.【Method】 A total of 282 pomelo accessions including landraces from different geographical regions and hybrid offspring of kiyomi tangor and pomelo were contained in this study. GBS library was constructed with genomic DNAs digested byR I restriction endonuclease and sequenced on Illumina HiSeq PE150 platform, the clean short reads were then mapped to pomelo reference genome by BWA, and SNPs were called out with SAMTOOLS pipeline. Based on 121 726 SNPs genotyping data, principal component analysis (PCA) and population genetic structure analysis were carried out and phylogenetic trees were constructed with Neighbor-joining method. Furthermore, two sub-populations containing high-acid accessions (32) and low-acid accessions (23) were used to identify candidate genes related to fruit acidity by Fst and XP-CLR selective sweeping analysis. Meanwhile, the genotype data of 282 pomelo accessions and the phenotypic data of titratable acid content in fruit were used for GWAS.【Result】A total of 201.66 Gb original reads were generated from 282 pomelo germplasm by GBS approach, in average each sample produced 0.72 Gb reads. After the screening conditions of sequencing depth of dp5, the miss less than 0.2 and minor alleles frequency (MAF)>0.01, and a total of 121 726 SNPs were selected out for subsequent analysis. The PCA, structure and phylogenetic analysis all supported that the 282 pomelo germplasm could be divided into 6 subgroups, among which pomelo and kiyomi hybrid population, grapefruits and other pomelo hybrid populations could be obviously different from true-to-type pomelo populations, pomelos originated from different geographical region displayed unique genetic feature. The pomelo germplasm from Thailand and Vietnam formed a relatively unique group different from other domestic groups, such as ShaTian pomelo, Wen Dan pomelo, and Dian Jiang pomelo in China. The genetic introgression from Vietnam pomelos were exhibited in most pomelo germplasm in southern China, suggested that Vietnam was the origin center for pomelo. In addition, some pomelo germplasm with unknown origin have been identified accurately by GBS technology. This study showed that different geographical distribution and artificial selection pressure had great effect on the genomic composition of pomelo. Besides, Fst, XP-CLR selective sweeping analysis revealed a strong selection signal region on chromosome 7 contained genes annotated as dihydrolipoyl transacetylase (DLT-E2) of pyruvate dehydrogenase complex (PDC) and aluminum-activated malate transporter (ALMT9), which involved in the synthesis and transportation of citric acid. In additional GWAS genome-wide association analysis identified another region on chromosome 2, which was also highly associated with fruit acidity. 【Conclusion】GBS technology provided reliable and efficient method for studying the phylogeny and evolution of pomelo. The study showed that artificial cross breeding, long-term artificial selection, geography isolation and domestication were the major driving forces for the formation of different types of pomelo germplasm. In addition, it clearly showed that Southeast Asian was primary center for pomelo origin and China mainland was secondary evolutionary center. Several candidate genes related to citric acid content in pomelo fruits were identified by Fst, XP-CLR selective sweeping and GWAS. This study provided important gene resources for the further genetic improvement and breeding of pomelo fruits.
GBS; pomelo phylogeny; genes related to fruit acidity; GWAS
10.3864/j.issn.0578-1752.2023.08.010
2022-05-25;
2022-10-08
国家重点研发计划(2018YFD1000101,2019YFD1001401)、种质资源精准鉴定(19211142)、江西科技计划项目(20161BBF60048)
通信作者江东,Tel:13983194771;E-mail:jiangdong@cric.cn
(责任编辑 赵伶俐)
猜你喜欢
中华诗词(2022年7期)2023-01-04 23:52:07
果树资源学报(2022年4期)2022-11-22 04:34:16
科学与财富(2022年9期)2022-07-02 13:43:04
中国果业信息(2021年12期)2021-12-02 00:38:04
寻根(2021年5期)2021-10-11 10:37:41
重庆与世界(2018年11期)2018-12-11 02:41:06
重庆行政(公共人物)(2018年2期)2018-06-28 09:50:22
现代园艺(2018年1期)2018-03-15 07:56:18
中国果业信息(2017年11期)2018-01-02 02:17:25
实用中医药杂志(2017年3期)2017-04-04 13:44:44
