
基于密度泛函理论和含时密度泛函理论下 富勒烯B24N24光谱性质研究
2023-04-29 02:32:46郭雅晶库雁兵李秀燕
四川大学学报(自然科学版) 2023年4期
郭雅晶 库雁兵 李秀燕


摘 要 :本文在B3LYP/6-31g(d)基组水平上,利用密度泛函理论(DFT)优化了B 24N 24团簇的几何和电子结构. 研究结果表明,优化所得B 24N 24团簇的几何结构对称性分别为S 4、C 2、S 8和O;在基态稳定结构基础上,得出其输运性质,既非p型输运材料,亦非n型输运材料,即不具有输运性质;在优化好的基态结构基础上,又研究了它的红外和拉曼分子振动谱,四种团簇的红外-拉曼振动谱的振动强度排序均为O>S 8>S 4>C 2;随后采用含时密度泛函理论(TDDFT),同样在B3LYP/6-31g(d)基组水平上,计算了这四种物质在CH 2Cl 2溶剂中的UV-Vis吸收谱特性,研究表明跃迁能级数量越多,吸收谱振动强度越强,并且这些分子的紫外-可见吸收谱的振动强度排序为S 4>C 2>S 8>O.
关键词 :含时密度泛函理论; 红外; 拉曼; 密度泛函理论
中图分类号 : O561 文献标识码 :A DOI : 10.19907/j.0490-6756.2023.045002
收稿日期: 2023-02-24
基金项目: 国家自然科学基金(11804246);山西省科技厅基础研究计划面上项目(202203021221214)
作者简介: 郭雅晶(1986-), 女, 山西太原人, 硕士, 高级实验师, 主要研究方向为凝聚态物理新型低维材料. E-mail: guoyajing58@163.com
DFT and TDDFT study on spectra of B 24N 24 fullerene
GUO Ya-Jing 1, KU Yan-Bing 2, LI Xiu-Yan 3
(1. Department of Physics, Taiyuan Normal University, Jinzhong 030619, China;
2. Shanxi Road and Bridge Construct Co., LTD., Lüliang 033000, China;
3. College of Physics and Optoelectronics, Taiyuan University of Technology, Taiyuan 030024, China)
The geometrical and electronic structure of B 24N 24 are optimized by using density functional theory (B3LYP) at the 6-31g(d) level. The results show that the geometrical structure symmetries of the optimized B 24N 24 clusters are S 4, C 2, S 8, O. Based on the stable structure of the ground state, it is concluded that its transport properties are neither the p-type transport material, nor the n-type transport material, so, they have no transport properties. On the basis of the optimized ground state structure, the IR-Raman spectra of the four clusters were obtained. And the order of vibrational intensity for the IR-Raman spectra of the four clusters is O>S 8>S 4>C 2. The Time-Dependent Density Functional Theory (TDDFT) was also used at the B3LYP/6 -31 g (d) level, the UV-Vis absorption spectra of these four compounds in CH 2Cl 2 were calculated, the results show that the more transition energy levels, the stronger the vibration intensity of the absorption spectrum. The vibrational intensities of the UV-Vis absorption spectra of these molecules are sorted as S 4>C 2>S 8>O.
Time-dependent density functional theory; Infrared; Raman; Density functional theory
1 引 言
由于富勒烯及相关类富勒烯纳米笼形结构具有潜在的电学、光学和磁学应用价值,人们对该类物质进行了深入研究 [1]. Radowan等 [2]对富勒烯团簇及其衍生物进行了研究.这引起了人们对(BN) n 类富勒烯结构特殊性质继续研究的兴趣. 与富勒烯C n 相比,(BN) n 具有良好的物理稳定性和化学惰性 [3]. 作为富勒烯C n 的等电子类似物,带隙能约为6 eV,且具有高能带、低温超导性、抗氧化性和较高的热力学稳定性等独特的物理化学性质 [4]. 除此以外,Stephan等人 [5]在纳米材料的电子辐射下合成了单层及嵌套的(BN) n ( n =12, 16, 28)物质;Oku 等人 [6]通过电弧熔融法和激光质谱法合成并检测了B 24N 24团簇. 同富勒烯C n 对比,氮化硼属于杂化富勒烯,它在构型方面遵循孤立四边形法则(ISR) [7]. 在笼形结构的(BN) n ( n >10)中,孤立四边形法则是指结构中的六个四元环完全被六元环分离,此时该物质具有相对较低能量且结构稳定 [8]. 通过理论计算,Wu等人 [9]研究发现,遵循孤立四边形法则的氮化硼团簇比对应的含有五边形和六边形的(BN) n 具有较高的稳定性. 该结论与Strout [10]和Stephan等人 [5]的研究结果一致.
Wu等人 [9]还根据密度泛函理论研究计算发现最稳定的B 24N 24类富勒烯结构的点群为S 8. 它由两个八边形、十六个六边形和八个四边形键合而成. 该团簇的结构稳定性高于仅由四元环和六元环构成的B 24N 24. 为了进一步研究B 24N 24团簇稳定结构的基态和激发态光谱性质,本文使用密度泛函理论(DFT)和含时密度泛函理论(TDDFT)量子化学计算方法,选取了富勒烯B 24N 24团簇的多种同分异构体为对比研究对象,通过TDDFT对大分子团簇在溶剂效应下进行计算,建立了适用于富勒烯(BN) n 的紫外可见吸收光谱,为较大体系笼形结构物质的光谱研究提供参考与借鉴.
2 计算细节
所有富勒烯结构采用Gaussian 09软件包 [11]在B3LYP/6-31g(d)基组水平 [12]上运用密度泛函理论(DFT)进行优化. 在考虑了不同的自旋多重性和大量的初始结构,还运用相同基组水平分析了振动频率,重点研究了B 24N 24四种稳定富勒烯结构 [13]. 在相同的理论水平上进行了谐波振动频率分析,并找出势能面上最小值,并且这里考虑的所有分子虚频值均为零(NIMAG=0) [14]. 基态分子的几何结构图像和分子轨道(MO)图像通过GaussView5.0 [15]程序生成.
为了进一步分析B 24N 24同分异构体的内部输运特征,在其基态结构的基础上,分别仿真计算了它们的电离势( IP )、电子亲和势( EA )、空穴/电子提取势( HEP/EEP )和空穴/电子重组能( λ h/ λ e,eV). 与此同时,在这些团簇稳定结构的基础上,还分别研究了它们的红外(IR)和拉曼(Raman)振动波谱,通过振动谱来分析它们的内部结构特征. 除此之外,还采用含时密度泛函理论(TD-DFT)结合极化连续介质模型(PCM)中的自洽反应场(SCRF)在CH 2Cl 2溶剂中来计算这四个同分异构体的紫外-可见吸收光谱(UV-Vis).
3 结果和讨论
3.1 几何结构
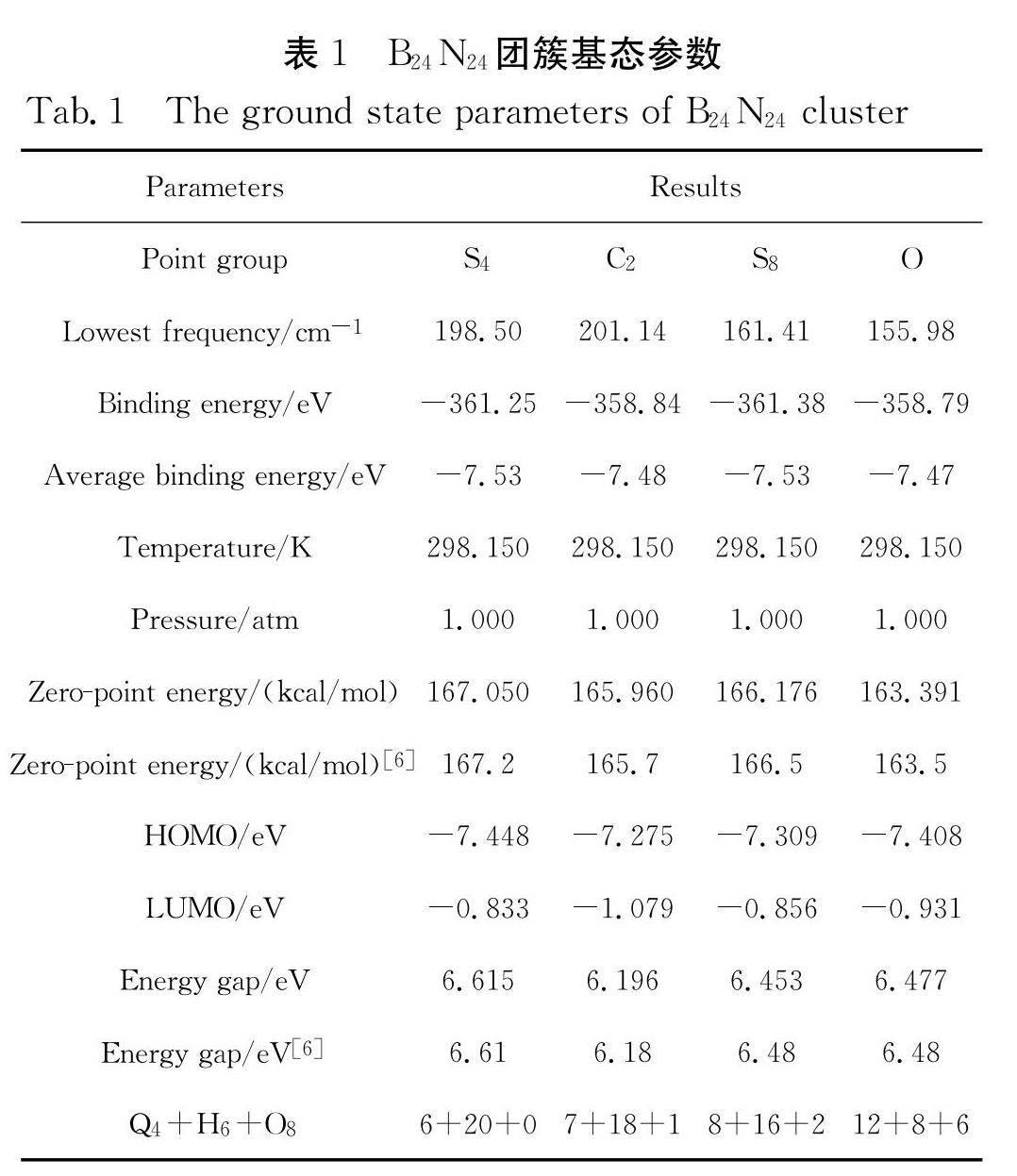
通过GaussView5.0构建了富勒烯B 24N 24多种点群结构,结合Gaussian 09程序在B3LYP/6-31g(d)基组水平上运用密度泛函理论(DFT)进行优化,并取得如图1所示四种稳定的基态结构. 从图1可以看出,粉色原子为硼原子(B),蓝色原子为氮原子(N),每个原子符号前的数字为设置分子结构时原子排列顺序数,且右下标S 4、C 2、S 8、O分别为各同分异构体的对称性(在下文中将分别用S 4、C 2、S 8、O代表四种团簇,用来区分这些同分异构体). 这四种结构分别由四边形、六边形和八边形键合而成. 考虑到孤立四边形法则,结合八边形参与其中构型的稳定性便会出现图1中的多种对称性稳定结构. 将图1与表1对照,表1中将四边形数量用Q 4表示,六边形的数量用H 6表示,八边形的数量则用O 8表示. 此外,表1还给出了这些同分异构体的基态对称性、最低频率、结合能、平均结合能、温度、压力、零点能、最高占据能HOMO、最低未被占据能LUMO和能隙.
从表1中得出,四种同分异构体最低频率均大于0 cm -1,则说明在该基组水平下计算所得最低频率都为正值,位于势能面的局部最小值,即NIMAG为0(无虚频). 再者,文献[6]的实验所得值与本文理论计算值对比可见,计算所得零点能167.050、165.960、166.176和163.391 eV与实验值167.2、165.7、166.5和163.5 eV非常接近,存在偏差是由于实验环境相较理论计算环境而言,总会存在外部细微干扰或不可抗因素. 表中的能隙值,实验所得为6.61、6.18、6.48和6.48 eV和计算值6.615、6.196、6.453和6.477 eV非常吻合,这进一步说明文中所选取优化基组水平是合理的. 对比表1中的四种同分异构体的结合能和平均结合能发现,拥有八个四边形、十六个六边形和两个八边形的对称性为S 8的B 24N 24最稳定,其次是具有六个四边形、二十个六边形对称性为S 4的B 24N 24团簇,排在最后的是具有十二个四边形、八个六边形、六个八边形对称性为O的B 24N 24分子,对称性为C 2的同分异构体B 24N 24分子则趋于S 4和O之间. 由此结果表明,对称性为S 8的B 24N 24最稳定,四种分子的稳定性排序为S 8>S 4>C 2>O.这与Wu等人 [9]的研究结果是一致的.
3.2 重组能
图2列出了富勒烯B 24N 24分子四种同分异构体的前线分子轨道,图中横坐标下的S 4、C 2、S 8、O分别为对应分子的对称性,纵坐标为HOMO、LUMO轨道所对应能级. 从图2中可看出,四中团簇的HOMO轨道主要执行π键轨道而LUMO轨道主要执行π *反键轨道,与此同时HOMO和LUMO能级π轨道特征以及离域的程度均在所有异构体的原子核. 所有异构体的HOMO轨道由N原子的p轨道形成p-π共轭,而B原子的p轨道几乎没有贡献;LUMO轨道由B原子的p轨道形成有效的p-π *共轭,而N原子的p轨道几乎没有贡献.
表2给出了这四种同分异构体的电离势( IP ,eV)、电子亲和能( EA ,eV)、空穴/电子提取势( HEP / EEP ,eV)和空穴/电子重组能量( λ h/ λ e,eV). 结合表1和表2可看出,共轭键的电离势和电子亲和能的变化与HOMO和LUMO能级的变化一致,如表1中B 24N 24同分异构体的HOMO能级大小顺序为S 4>O>S 8>C 2,并伴随有电离势随其顺序反向增加;而同分异构体的LUMO能级大小顺序为C 2>O>S 8>S 4,电子亲和能亦随其顺序反向增加. 由此可见,载流子的传输与前线分子轨道的分布密切相关,一般来讲,p型输运材料的 IP A在5.680~6.786 eV范围内,n型输运材料 EA A在2.411~3.141 eV范围内, IP A和 EA A材料的双极性传递应在5.905~7.026 eV和2.797~3.479 eV范围内. 由上述结果讨论可得出,B 24N 24的四类同分异构体既非p型输运材料、亦非n型输运材料,也不具有双极性传递性质.
3.3 红外和拉曼振动谱
在优化基态结构的基础上,在相同基组水平(B3LYP/6-31g(d))下分别计算了这四种同分异构体的红外和拉曼振动谱. 图3给出了富勒烯 B 24N 24同分异构体的红外和拉曼光谱,对称性为S 4的 B 24N 24团簇对应黑色光谱,对称性为C 2的B 24N 24团簇对应红色光谱,对称性为S 8的B 24N 24团簇对应绿色光谱,对称性为O的B 24N 24团簇对应橙色光谱.
从红外光谱可以看出,这四种分子的振动频率主要集中在750~900 cm -1和1350~1550 cm -1之间,900~1350 cm -1中间有少数零星散发的振动峰,振动强度弱;这四种同分异构体的振动最强峰均在1500 cm -1处,四个团簇相比,对称性为O的B 24N 24在该出的振动峰最强,其次是对应绿色波谱的S 8,然后是对应黑色波谱的S 4,最后是对应红色波谱的C 2. 由图3中的拉曼波谱看出,各分子的拉曼振动峰遍布100~1600 cm -1之间,比各自相对应的红外振动波峰数量多;所有分子的两个最强拉曼振动峰集中在400~500 cm -1和800~900 cm -1之间,并且橙色振动谱对称性为O的B 24N 24在这两处的振动强度最大,其次是对应绿色波谱的S 8,然后是对应黑色波谱的S 4,最后是对应红色波谱的C 2.这与红外振动谱的研究结果相同.
由上述研究得出,红外吸收光谱的波数在500~ 1600 cm -1之间,属于中红外区(400~4000 cm -1,2.5~25 μm),即属于基本振动区;这四种团簇的红外光谱振动强度排序为O>S 8>S 4>C 2. 对于拉曼光谱,这四种同分异构体的振动强度排序亦为O>S 8>S 4>C 2. 由于这四种团簇的构型为非线性分子,故这些分子的振动自由度为3 n -6个( n 为原子数).
3.4 紫外-可见吸收谱
运用含时密度泛函理论(TDDFT),结合极化连续介质模型(PCM)中的自洽反应场(SCRF)在原有基组水平(B3LYP/6-31g(d))上计算了四种同分异构体溶于CH 2Cl 2溶剂的紫外可见吸收谱(UV-Vis). 图4给出了四中团簇在CH 2Cl 2溶剂中的吸收谱,点群为S 4的B 24N 24对应黑色吸收谱,点群为C 2的B 24N 24对应红色吸收谱,点群为S 8的B 24N 24对应绿色吸收谱,点群为O的B 24N 24对应橙色吸收谱. 表3给出了所有分子在CH 2Cl 2溶剂中的吸收特性,同时列出了部分主要能级跃迁和占比百分以及对应波长和振动强度;表中列出了相应峰值的能量跃迁出处,例如图4中S 4在202.30 nm最高吸收峰吸收能量主要是由于HOMO-5→LUMO (36%)、HOMO-2→LUMO+1(10%)和HOMO→LUMO+4(22.4%)的轨道电子跃迁产生.
由图4可以看出,B 24N 24分子的吸收峰波长集中在175~270 nm之间,并且都只有一个最强吸收峰,四个团簇相比对称性为S 4的吸收峰振动强度最强,其次是C 2,然后是S 8,最后是O;可见所有团簇的吸收谱波长均在紫外波长范围内(10~400 nm) [16],处于高频短波段UVC(100~280 nm) [17],而且处于灭 菌紫外线(200~280 nm) [18]波段和制臭氧紫外线(100~200 nm) [19]波段. 结合表3可得,跃迁能级数量越多,吸收谱振动强度越强,例如S 8与O相比,S 8的吸收峰振动强度比O吸收峰强,并且贡献于S 8吸收峰的跃迁能级数量比O的多;同样以S 4和S 8相对比,S 4的吸收峰振动强度之所以比S 8强,是由于作用于S 4吸收峰的跃迁能级数量多于S 8;所有分子的紫外-可见吸收谱的振动强度排序为S 4>C 2>S 8>O.
4 结 论
基于密度泛函理论(DFT)在B3LYP/6-31g(d) 基组水平上,研究了优化后的类富勒烯B 24N 24四种同分异构体的基态性能(点群、结合能、虚频、能隙、重组能和红外-拉曼振动谱等). 结果表明,四种B 24N 24团簇基态结构对称性分别为S 4(由6个四边形,20个六边形构成)、C 2(由7个四边形,18个六边形,1个八边形构成)、S 8(由8个四边形,16个六边形,2个八边形构成)和O(由12个四边形,8个六边形,16个八边形构成);B 24N 24的四类同分异构体既非p型输运材料、亦非n型输运材料,也不具有双极性传递性质;四种团簇的红外-拉曼振动谱的振动强度排序均为O>S 8>S 4>C 2. 随后采用含时密度泛函理论(TDDFT)同样在B3LYP/6-31g(d)基组水平上,计算了这四种物质在CH 2Cl 2溶剂中的UV-Vis吸收谱特性,研究表明跃迁能级数量越多,吸收谱振动强度越强;并且这些分子的紫外-可见吸收谱的振动强度排序为S 4>C 2> S 8>O.
参考文献:
[1] Wang Y, Yao M G, Hua X, et al . Structural evolution of D 5h(1)-C 90 under high pressure: A mediate allotrope of nanocarbon from zero-dimensional fullerene to one-dimensional nanotube[J]. Chin Phys Lett, 2022, 39: 056101.
[2] Radowan A, Elmorsy K, Hassan A H, et al . Manganese dioxide (MnO 2)/fullerene-C 60-modified electrodes for the voltammetric determination of rifaximin [J]. J Anal Test, 2021, 5: 341.
[3] Xie G Y, Lv H, Chen X, et al . The discovery structure properties and application of fullerene C 60 [J]. Carbon, 2021, 3: 34.
[4] Goyal R N, Gupta V K, Bachheti N. Fullerene-C 60-modified electrode as a sensitive voltammetric sensor for detection of nandrolone-an anabolic steroid used in doping [J]. Anal Chim Acta, 2007, 597: 82.
[5] Stephan O, Bando Y. Atomic structure and electronic state of boron nitride fullerenes and nanotubes[J]. Appl Phys A, 1998, 67: 107.
[6] Oku T, Kuno M, Narita I. Structure and stability of perazido substituted azacycloalkanes [J]. Diamond Relat Mater, 2002, 11: 940.
[7] Orfanopoulos M, Kambourakis S. Fullerene C 60 and C 70 photosensitized oxygenation of olefins [J]. Tetrahedron Lett, 2014, 35: 1945.
[8] Zope R R, Dunlap B I. Are hemispherical caps of boron-nitride nanotubes possible? [J]. Chem Phys Lett, 2004, 386: 403.
[9] Wu H S, Xu X H, Zhang F Q, et al . What is the most stable B 24N 24 fullerene? [J]. J Phys Chem A, 2003, 107: 6609.
[10] Strout D L. Structure of boron nitride E-phase: diamond lattice of B 12N 12 fullerenes [J]. Chem Phys Lett, 2004, 95: 383.
[11] Frisch M J, Trucks G W, Schlegel H B, et al . Gaussian 09 [M]. Revision A.02. Wallingford CT: Gaussian Inc., 2009.
[12] Frisch A, Nielsen AB, Holder A J. Gaussview user manual [M]. Pittsburgh PA: Gaussian Inc., 2005.
[13] Rassolov V A, Ratner M A, Pople J A, et al . 6-31G * Basis set for third-row atoms [J]. J Comput Chem, 2001, 22: 976.
[14] Rassolov V A, Pople J A, Ratner M A, et al . 6-31G * basis set for atoms K through Zn [J]. J Chem Phys, 1998, 109: 1223.
[15] Frisch A, Nielsen A B, Holder A J. Gaussview 5.0 [M]. Pittsburgh PA: Gaussian Inc., 2005.
[16] Runge E, Gross E K U. Density-functional theory for time-dependent systems [J]. Phys Rev Lett, 1984, 52: 997.
[17] Wang J L, Zhou X L, Wang G H, et al . Optimally stuffed fullerene structures of silicon nanoclusters [J]. Phys Rev B, 2005, 71: 113412.
[18] Tian W Q, Chen D L, Cui Y H, et al . Structures, stabilities, electronic and optical properties of fullerenes (C 50-C 68) and derivatives [J]. Comput Theor Nanosci, 2009, 6: 239.
[19] Zope R R, Baruah T, Pederson M R, et al . Theoretical infrared, Raman, and optical spetra of the B 36N 36 cage [J]. Phys Rev A, 2005, 71: 025201.
猜你喜欢
风流一代·经典文摘(2023年5期)2023-05-21 11:42:11
环球时报(2022-05-23)2022-05-23 11:28:37
辽宁省博物馆馆刊(2021年0期)2021-07-23 07:25:36
金桥(2021年4期)2021-05-21 08:19:20
电子制作(2019年7期)2019-04-25 13:18:24
电子制作(2019年7期)2019-04-25 13:17:14
电子测试(2018年18期)2018-11-14 02:30:36
电子制作(2018年14期)2018-08-21 01:38:40
光学精密工程(2016年3期)2016-11-07 09:03:43
光学精密工程(2016年1期)2016-11-07 09:01:00
