
基于EF-1α和β微管蛋白基因序列的棉花枯萎病菌遗传多样性和单倍型分析
2023-04-29 08:47王海莹陈小海钟烨仪龚举武刘平ChinYaoxian王沛政袁有禄
棉花学报 2023年4期
关键词:棉花
王海莹 陈小海 钟烨仪 龚举武 刘平 Chin Yaoxian 王沛政 袁有禄


摘要:【目的】通過分析棉花枯萎病菌的遗传多样性,探究新疆棉花枯萎病菌株的分群及其演化。【方法】2022年在新疆不同植棉区共分离出22株棉花枯萎病菌株,对延伸因子1α(elongation factor-1α, EF-1α)和β微管蛋白基因进行扩增、测序,并从美国国立生物技术信息中心(National Center for Biotechnology Information, NCBI)数据库获取36个棉花枯萎病菌株的相关基因序列信息。基于上述基因序列分别进行系统进化分析和单倍型分析。【结果】基于57条EF-1α基因序列的进化树分析表明,棉花枯萎病菌可分为3大群,第1大群包含来自新疆、河北和澳大利亚的共31个枯萎病菌株,该大群可分成4个亚群;第2大群包含25个枯萎病菌株,构成比较复杂,可分成3个亚群;第3大群仅包含美国菌株LA140。基于28条β微管蛋白基因序列的进化树分析表明,本次分离的新疆棉花枯萎病菌株与棉花枯萎病菌7号和8号生理小种不同。根据EF-1α基因序列构建的单倍型网络将棉花枯萎病菌株分为19个单倍型,新疆21个棉花枯萎病菌株归属于有共同起源的5种单倍型。【结论】本研究分离的新疆棉花枯萎病菌株与已报道的棉花枯萎病菌1~8号生理小种均不相同,但与河北菌株的亲缘关系较近。EF-1α单倍型分析表明,本研究中的所有棉花枯萎病菌均从1号生理小种演化而来。
关键词:棉花;枯萎病菌;单倍型分析;遗传多样性分析;延伸因子1α;β微管蛋白
Genetic diversity and haplotype analysis of cotton Fusarium wilt based on gene sequences of EF-1α and β-tubulin
Wang Haiying1, 2, Chen Xiaohai2, 3, Zhong Yeyi2, 3, Gong Juwu1, Liu Ping1, Chin Yaoxian2, 3, Wang Peizheng2, 3*, Yuan Youlu1
(1. Institute of Cotton Research of Chinese Academy of Agricultural Sciences/National Key Laboratory of Cotton Bio-breeding and Integrated Utilization, Anyang, Henan 455000, China; 2. Key Laboratory for Coastal Marine Eco-environment Process and Carbon Sink of Hainan Province, Hainan Tropical Ocean University, Sanya, Hainan 572022, China; 3. Yazhou Bay Innovation Institute, Hainan Tropical Ocean University, Sanya, Hainan 572022, China)
Abstract: [Objective] This research aims to characterize the grouping and evolution of cotton Fusarium oxysporium f. sp. vasinfectum (FOV) strains in Xinjiang on the basis of genetic diversity analysis. [Method] A total of twenty-two FOV strains from different cotton planting areas in Xinjiang were isolated in 2022, and EF-1α and β-tubulin gene were amplified and sequenced. Sequences of other thirty-six cotton FOV strains were downloaded from National Center for Biotechnology Information (NCBI). Phylogenetic analysis and haplotype analysis were carried out based on the above-mentioned sequences. [Result] Phylogenetic tree analysis based on fifty-seven EF-1α gene sequences indicated that FOV strains can be divided into three groups. The first group included 31 FOV strains from Xinjiang, Hebei province and Australia. This group can be further divided into four subgroups. The second group, including 25 FOV strains and relatively complex composition, can be divided into three subgroups. The last group only included LA140 from America. Phylogenetic tree analysis based on twenty-eight β-tubulin gene sequences showed that FOV strains from Xinjiang are different from the race 7 and race 8. All strains were divided into nineteen haplotypes by haplotype network analysis based on EF-1α sequences. Twenty-one FOV strains isolated from Xinjiang belongs to five haplotypes, which share a common origin. [Conclusion] The strains isolated from Xinjiang in this research are different from the reported race 1 to race 8, but share a relatively close relationship with FOV in Hebei province. Haplotype analysis of EF-1α indicates all FOV strains in this research are evolved from race 1.
Keywords: cotton; Fusarium wilt; haplotype analysis; genetic diversity analysis; EF-1α; β-tubulin
棉花枯萎病是1种危害性极大的世界性土传病害。棉花枯萎病的致病真菌是尖孢镰刀菌萎蔫专化型(Fusarium oxysporium f. sp. vasinfectum),受其侵染的棉花茎秆维管束坏死,进一步导致棉株萎蔫甚至枯死。棉花枯萎病的普遍发生和扩展,已对棉花生产的持续发展构成了严重威胁,并且给植棉区造成巨大的经济损失。棉花枯萎病菌毒力强、分布广、危害严重,但其内部菌系也存在明显的致病性分化。了解棉花枯萎病菌的分类、起源进化等,可对该病原菌今后的进化趋势做出预测,为棉花枯萎病抗病育种和综合防治提供理论依据。
自20世纪60年代以来,国内外学者对棉花枯萎病菌的生理生化特性开展了大量研究。研究人员借助海岛棉(Gossypium barbadense)、陆地棉(G. hirsutum)、亚洲棉(G. arboreum)、烟草(Nicotiana tabacum)等鉴别寄主,将棉花枯萎病菌分为8个生理小种[1-3]。棉花枯萎病菌1号、2号、3号、4号和8号生理小种在美国有分布[4-5]。Bridge等[6]发现棉花枯萎病菌1号和2号生理小种主要分布在美国和坦桑尼亚,且这2个生理小种的致病性较强。Davis等[7]研究发现棉花枯萎病菌3号生理小种分布在埃及、以色列、苏丹和中国。4号生理小种最初在印度被发现,并且很快成为美国加利福尼亚州棉花枯萎病的优势生理小种[3]。5号生理小种最先在苏丹被发现,并根据分子分类方法和致病性测试将3号和5号小种归为同一生理小种。6号生理小种分布在巴西和巴拉圭。7号生理小种主要分布在中国[8]。研究人员通过分子标记等技术,将4号和7号小种归为同一生理小种[9]。专家学者借助人工接种鉴别寄主的方法,发现我国存在棉花枯萎病菌3号、7号和8号生理小种,其中7号生理小种为优势生理小种,而3号和8号生理小种少量分布于内地[1]。
依据病原菌的遗传多样性和遗传距离研究可以初步判断其所属的生理小种类型,并为植物病害防控策略及育种计划的制定提供有效参考,国内外学者在这方面已开展了大量研究。Abd-Elsalam等[10]利用随机扩增多态性(random amplified polymorphic DNA, RAPD)和扩增片段长度多态性(amplification fragment length polymorphisms, AFLP)标记,以棉花枯萎病菌1号、2号、3号、4号和5号生理小种等为参考菌株,对来自埃及的棉花枯萎病菌进行了研究,结果表明埃及棉花的枯萎病菌可能为3号生理小种,且与5号生理小种亲缘关系较近,而与1号生理小种亲缘关系较远。Assigbetse等[11]利用RAPD分子标记技术将棉花枯萎病菌分为3个群,其中1号、2号和6号生理小种属于1群,4号生理小种属于2群,3号和5号生理小种属于3群。Cianchetta等[12]利用翻译延伸因子(elongation factor-1α, EF-1α)基因的部分序列有效鉴定了棉花枯萎病菌1号、2号、3号、4号和8号生理小种,推测棉花枯萎病菌可能有2个起源群。田新莉等[13]利用RAPD标记将新疆棉花枯萎病菌的32个代表菌株划分为5个群,认为新疆棉花枯萎病菌群体以7号生理小种为主。白剑宇[14]通过营养亲和群和指纹图谱扩增技术,将新疆棉花枯萎病菌划分为3个类群,第1类群包括7号生理小种及其亲和菌株,是新疆的优势生理小种,但不包括3号、8号生理小种;第2和第3类群与3号、7号、8号生理小种均不亲和。陈文霞[15]利用AFLP分子标记将新疆棉花枯萎病菌划分为分别以3号、7号、8号等生理小种为代表的3個群,并认为新疆棉花枯萎病菌的遗传变异不大。Guo等[16]利用AFLP标记研究发现我国棉花枯萎病菌以7号生理小种为主,还发现了不同于3号、7号、8号等生理小种的新生理型菌株。郭庆港等[8]利用EF-1α基因序列将河北省主要植棉区的77株枯萎病菌分为5个大群,这5个群来自2个起源,与澳大利亚生理型菌株具有较近的亲缘关系。尽管与尖孢镰刀菌种内遗传多态性相关的研究报道较多,但目前对尖孢镰刀菌生理小种的起源与进化仍然知之甚少。
研究者多利用持家基因序列,如EF-1α和β-微管蛋白(β-tubulin),对棉花枯萎病菌等植物病原菌的遗传多样性进行分析[17]。EF-1α普遍参与核糖体蛋白的翻译,在种内具有较高的保守性,其基因序列被广泛应用于真菌物种的鉴定分析,且相关序列信息查询方便,易于不同研究结果间的相互比较[12]。β-tubulin是细胞内的一种骨架蛋白,是微管结构最主要的组成成分之一,在真核生物中具有高度保守性,参与维持细胞形态和有丝分裂等[18]。
单倍体基因型(单倍型)是指共存与单条染色体上的一系列遗传变异位点的组合。由多个单核苷酸多态性(single nucleotide polymorphism, SNP)突变构成的1种突变谱可被称为1种单倍型。单倍型分析技术是寻找单条染色体上杂合SNP变异位点的有效方法[19]。在群体遗传学上可用于分析等位基因的变异,追踪个体间的亲缘关系,了解生物的进化历史等[20-21]。单倍型分析技术近年来也不断向菌物学方向渗透。李杨等[22]通过单倍型分析技术揭示油茶炭疽病原菌暹罗刺盘孢菌(Colletotrichum siamense)具有丰富的遗传多样性,为油茶炭疽病害的防治提供了可靠依据。夏块菌(Tuber aestivum)是1种可食用的子实体,在全球广受欢迎,Riccioni等[23]通过构建单倍型网格分析对夏块菌的多样性保护提出了针对性意见。
目前为止,关于利用基因序列分析新疆棉花枯萎病菌的遗传多样性和起源进化的研究鲜有报道。本研究通过对2022年新疆不同植棉区棉花枯萎病菌菌株的分离、EF-1α和β-tubulin基因扩增和测序,并在此基础上进行棉花枯萎病菌的遗传多样性和单倍型分析,从而初步判断供试菌株的遗传分群和演化等,为今后新疆植棉区棉花枯萎病菌的分类、起源进化研究和综合防控等提供科学依据。
1 材料与方法
1.1 试验材料
2022年3月对呈现棉花枯萎病症状的茎秆进行棉花枯萎病菌的分离、单孢纯化。具体方法为:将茎秆剪成长度为1 cm的小段,剥下表皮,用酒精消毒后放入超净台中0.1%的升汞水溶液中杀菌30 s,无菌水清洗3次后接入察氏培养基中,25 ℃培养3~4 d即可长出大量白色菌丝。采用平板划线法进行枯萎病菌单孢分离纯化,获得新疆棉花枯萎病菌菌株。
茎秆材料来源如下:新疆博乐市达镇红坡4队(以下简称达镇)2株(DAZHEN 6和DAZHEN 20);新疆生产建设兵团第八师一四九团十三连(以下简称13连)6株(13LIAN 8、13LIAN C1、13LIAN X11、13LIAN X30、13LIAN X31和13LIAN C17);新疆生产建设兵团第六师芳草湖农场三场(以下简称三场)1株(SANCHANG C21);新疆生产建设兵团第七师125团1连(以下简称奎屯)1株(KUITUN C23);三亚师部农场新疆南繁基地(以下简称师部)12株(SHIBU C2、SHIBU C3、SHIBU C4、SHIBU X12、SHIBU X13、SHIBU X14、SHIBU W2、SHIBU W3、SHIBU W4、SHUBU W6、SHIBU W9和SHIBU V2)。
另外36个棉花枯萎病菌株的基本信息和EF-1α、β-tubulin基因参考序列通过美国国立生物技术信息中心(National Center for Biotechnology Information, NCBI)數据库查询并下载(表1)。
1.2 EF-1α和β-tubulin基因序列扩增
首先将分离纯化的枯萎病菌株加入到含有100 mL察氏液体培养基的锥形瓶中,于摇床(25 ℃,180 r·min-1)中振荡培养5 d,吸取1.5 mL菌液放入无菌的1.5 mL离心管中,10 000 r·min-1离心30 s,尽可能吸干净上清液,收集菌体。采用植物基因组DNA提取试剂盒(通用型,广州擎科生物技术有限公司)提取菌体DNA,菌体DNA提取的详细操作步骤参见试剂盒说明书。DNA提取完成后,使用NanoGenius超微量光度计(上海美谱达仪器有限公司)检测DNA原液的浓度。
扩增EF-1α和β-tubulin基因序列的引物序列如下。EF-F:5'-CACCTTAACGTCGTCGTCATC-3',EF-R:5'-GGAAGTACCAGTGATCATGTT-3';β-tubulin-F:5'-CGTCTAGAGGTACCCATACCG-GCA-3',β-tubulin-R:5'-GCTCTAGACTGCTTT-CTGGCAGACC-3'[24]。PCR扩增试剂盒购自广州擎科生物技术有限公司。PCR反应体系:模板 DNA(20~80 ng·μL-1)2.0 μL,2×PCR Mix 10.0 μL,上下游引物(10 μmol·L-1)各1.0 μL,灭菌超纯水6.0 μL。PCR反应程序如下:97 ℃ 1 min;96 ℃ 30 s,50 ℃ 1 min,72 ℃ 1 min,37个循环;72 ℃ 10 min。扩增产物送往广州擎科生物技术有限公司进行测序。
1.3 棉花枯萎病菌遗传多样性和单倍型分析
利用MEGA 7软件选择邻近法(neighborjoining, NJ)分别构建基于EF-1α和β-tubulin基因序列的系统进化树,自展值(Bootstrap值)设置为1 000。借助MEGA 7软件进行序列比对,利用DnaSP v6软件分析基因序列的多态性,获得序列的保守位点数、变异位点数、单一变异位点数、单倍型个数、单倍型多态性、核苷酸多样性等信息。使用Network软件绘制单倍型网络图。
2 结果与分析
2.1 棉花枯萎病菌遗传多样性分析
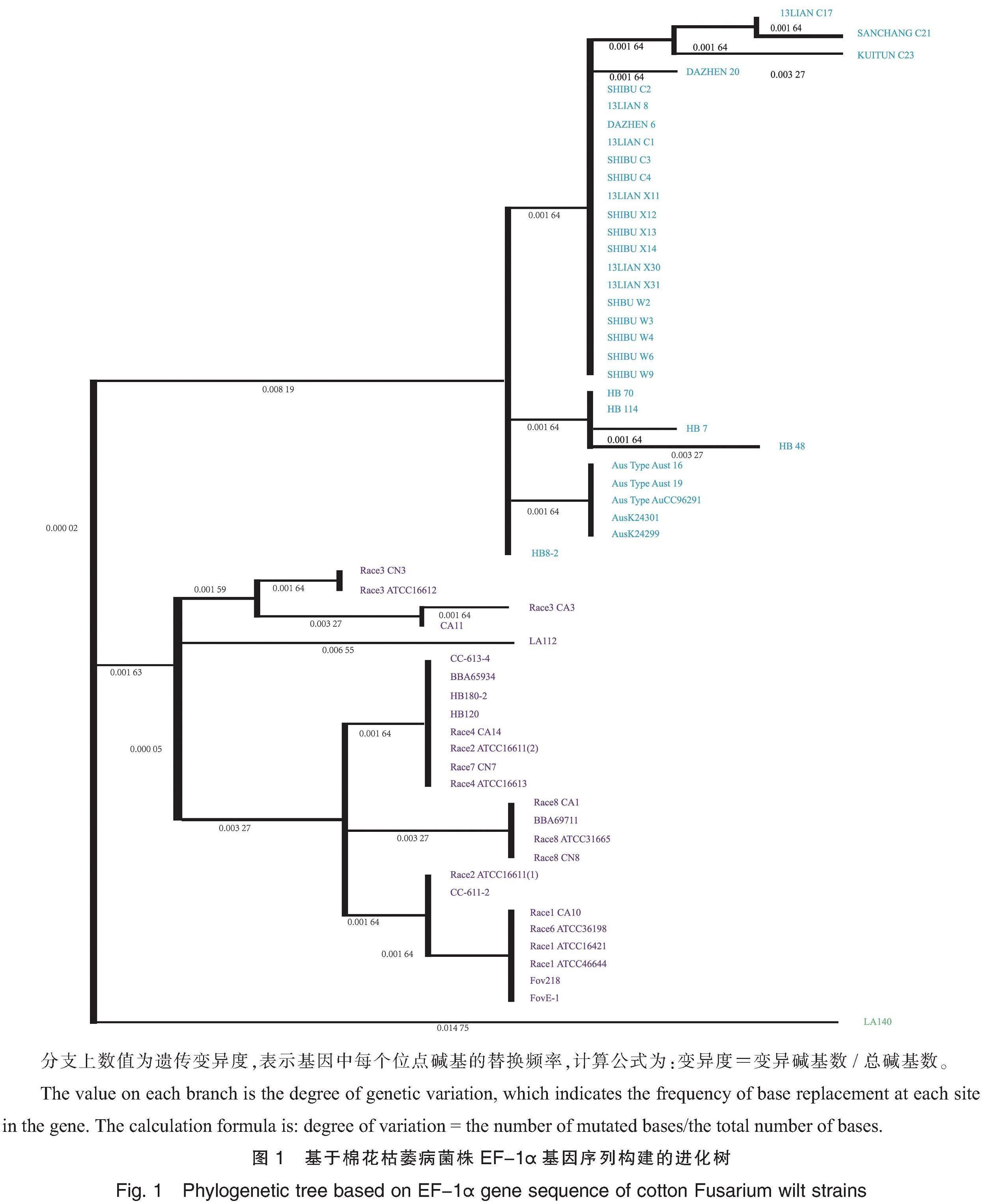
本研究中新疆棉花枯萎病菌22个供试菌株共来源于5处,其中4处在新疆(共10株),另1处为新疆北疆的繁育基地(海南三亚,共12株)。通过菌株的分离、鉴定和基因测序得到EF-1α基因的21条序列信息(其中SHIBU V2菌株未扩增出条带),其余36条EF-1α基因序列通过文献查找并从GenBank下载获得。基于上述57条EF-1α基因序列(长度为611 bp)构建系统进化树,显示各分支的遗传变异程度。结果表明,棉花枯萎病菌株可分成3大群(图1)。第1大群为来自新疆、河北和澳大利亚的枯萎病菌株,该群又可以分成4个亚群,第1亚群由来自新疆的21个枯萎病菌株构成,第2亚群由来自河北的4个枯萎病菌株构成,第3亚群由来自澳大利亚的5个枯萎病菌株构成,第4亚群包括来自河北的1个枯萎病菌株(HB8-2),这4个亚群的亲缘关系较近。第2大群包含25个菌株,该大群又可以分成3个亚群,第1亚群主要包含棉花枯萎病菌3号生理小种(共4株),第2亚群包含1个菌株LA112,第3亚群(共20株)主要包括棉花枯萎病菌1号、2号、4号、6号、7号、8号生理小种和来自河北的棉花枯萎病菌株(HB180-2和HB120)等。第3大群只有1个来自美国的菌株LA140。根据本次对EF-1α基因序列的分析结果,新疆棉花枯萎病菌株和河北枯萎病菌株等在内的第1大群与其他2个大群的亲缘关系较远。本研究表明,基于EF-1α基因序列,新疆棉花枯萎病菌株与已报道的棉花枯萎病菌1~8号生理小种均存在差异。
基于β-tubulin基因序列构建的进化树见图2。该进化树中共包含28条序列,其中5条序列(长度为370 bp)是本研究中从新疆菌株SHIBU W6、SHIBU W9、SHIBU X14、SHIBU V2、SHIBU W2中扩增测序所得(其他材料未扩增剂基因序列),另外23条序列是通过文献查找不同来源的棉花枯萎病菌生理小种(菌株)[25],并从GenBank下载获得。结果表明,以上棉花枯萎病菌株可以分为3大群,第1大群(共17株)包括本研究中分离的新疆5个菌株和1号、2号、3号、4号、6号、8号生理小种以及来自澳大利亚的菌株;第2大群(共10株)包括来自河北(7株)、中国的3号与8号枯萎病菌生理小种(各1株)和来自澳大利亚的菌株(1株);第3大群,仅1株,为中国的7号生理小种。
2.2 棉花枯萎病菌株EF-1α和β-tubulin基因的单倍型分析
基于上述棉花枯萎病菌EF-1α基因的57条序列和β-tubulin基因的28条序列进行了单倍型分析,结果(表2)表明棉花枯萎病菌EF-1α基因可分为19个单倍型,β-tubulin基因可分为9个单倍型,EF-1α基因的单倍型多态性较β-tubulin基因更高,但其核苷酸多样性低于β-tubulin基因。
基于EF-1α基因序列构建的单倍型网络图见图3,各单倍型对应的菌株信息见表3。EF-1α基因单倍型网络图表明,棉花枯萎病菌单倍型均由H8(1号生理小种)单倍型演化而来。以H8单倍型作为棉花枯萎病菌进化演化路线来看,共有5条演化路线。第1条演化路线包括单倍型H1~H5(本研究分离的新疆菌株)和H12(河北菌株)~H16(澳大利亚菌株)。第2条演化路线包括单倍型H6~H7(包含1号、2号、3号、4号、6号、7号和8号生理小种)和H9~H10(包含2号、3号、4号和8号生理小种、澳大利亚菌株和河北菌株)。其他3条演化路线包含的菌株数很少,分别为H11和H17、H18、H19单倍型。结果表明,根据EF-1α基因序列,新疆21个枯萎病菌株中有17个菌株属于H1单倍型,其余4个菌株分别属于H2~H5单倍型。其他多种类型的生理小种和部分澳大利亚枯萎病菌株属于H6~H9单倍型。河北枯萎病菌株属于H10~H12单倍型。其他部分澳大利亚枯萎病菌株属于H15~H16单倍型。BBA65934菌株属于H17單倍型。埃及枯萎病菌株FovE-1单独属于H18单倍型。BBA69711菌株属于H19单倍型。
由于扩增出的β-tubulin基因序列与我国的3号和7号生理小种基因序列差异较大,所以在单倍型网络构建过程中将 Race3 CN3 和Race7 CN7的β-tubulin基因序列删去。基于26条β-tubulin基因序列构建的单倍型网络图见图4,各单倍型对应的菌株信息见表4。结果表明,单倍型H7~H9分到1个类群,主要包括来自河北的菌株和1个8号生理小种菌株(Race8 CN8)及1个澳大利亚菌株(Aus Type ATCC96291)等。其余6个单倍型(H1~H6)归为第2类群,以单倍型H1为核心分别演化出H2~H6等单倍型,其中单倍型H1主要包括本研究分离的5个新疆棉花枯萎病菌株和1个1号生理小种菌株(Race1 ATCC16421)。
3 讨论
真核生物延伸因子EF-1α在蛋白质翻译过程中发挥重要作用,由于在种内具有较高的保守性,被广泛应用于真菌物种的鉴定分析[26]。本研究利用新疆不同植棉区的棉花枯萎病菌21条EF-1α基因序列和GeneBank数据库中的36条EF-1α基因序列构建进化树,将57个棉花枯萎病菌株分成3大群,其中新疆棉花枯萎病菌株与河北及澳大利亚的枯萎病菌株亲缘关系较近,与其他报道的棉花枯萎病菌株亲缘关系较远,包括先前国内报道的棉花枯萎病菌7号和8号生理小种。这与郭庆港等[8]报道的河北省主要植棉区的枯萎病菌与澳大利亚生理型菌株具有较近的亲缘关系,而与其他枯萎病菌株亲缘关系较远相符。但本研究发现新疆棉花枯萎病菌株与河北的枯萎病菌株存在差异,这与白剑宇[14]、陈文霞[15]报道的新疆棉花枯萎病菌以7号小种为优势生理小种的研究结果不同。本研究中分离的新疆棉花枯萎病菌株与河北菌株及澳大利亚菌株亲缘关系较近,表明新疆棉花枯萎病菌的来源可能与早期新疆从国内其他省份频繁引进棉花品种有关。本研究中基于57条EF-1α基因序列构建的进化树将棉花枯萎病菌株分成3大群,与Assigbetse等[11]、白剑宇[14]、陈文霞[15]将棉花枯萎病菌分为3个群的结果类似,而与田新莉等[13]将新疆棉花枯萎病菌32个代表菌株划分为5个群的结果不同。同时基于28条β-tubulin基因序列构建的进化树也将棉花枯萎病菌株分为3大群。本研究将棉花枯萎病菌3号和5号生理小种归为同一生理型,与Skovgaard等[9]的研究报道相同。
基于棉花枯萎病菌株57条EF-1α基因序列进行的单倍型分析表明,棉花枯萎病菌共分为19个单倍型。新疆棉花枯萎病菌株属于单倍型H1~H5。本研究中所有棉花枯萎病菌的单倍型均由H8单倍型(枯萎病菌1号生理小种)演化而来。基于棉花枯萎病菌株28条β-tubulin基因序列进行的单倍型分析表明,棉花枯萎病菌可分为9种单倍型。以单倍型H1为核心分别演化出H2~H6等单倍型,其中单倍型H1主要包括本研究分离的新疆棉花枯萎病菌株和枯萎病菌1号生理小种。以上结果也表明棉花枯萎病菌单倍型可能最早也由棉花枯萎病菌1号生理小种演化而来,这与棉花枯萎病菌1号生理小种最早在美国被发现,并在美国植棉区广泛分布的报道相吻合[27]。而Skovgaard等[9]利用分子系统学研究表明棉花8个枯萎病菌生理小种至少有2个独立的进化起源;郭庆港等[8]和Cianchetta等[12]认为棉花枯萎病菌可能来自2个起源群。
4 结论
基于棉花枯萎病菌EF-1α基因序列的遗传多样性,将棉花枯萎病菌株分成3大群。第1大群包含本次分离出、来自河北和澳大利亚的共31个菌株;第2大群包含25个菌株,可以分成4个亚群;第3大群仅有来自美国的菌株LA140。第1大群与其他2大群的亲缘关系较远,表明本次分离出的新疆棉花枯萎病菌与已报道的棉花枯萎病菌1~8号生理小种均不相同,但与河北分离的枯萎病菌亲缘关系较近。β-tubulin基因进化树分析也表明本次分离的新疆棉花枯萎病菌株与7号和8号生理小种不同。基于EF-1α基因序列构建的单倍型网络将棉花枯萎病菌株分为19个单倍型,新疆21个棉花枯萎病菌株有共同的起源,属于H1~H5单倍型,本研究中的所有棉花枯萎病菌均由1号生理小种演化而来。
参考文献:
[1] 陈其煐, 籍秀琴, 孙文姬. 我国棉枯萎镰刀菌生理小种研究[J]. 中国农业科学, 1985, 18(6): 1-6.
Chen Qiying, Ji Xiuqin, Sun Wenji. Identification of races of cotton-wilt Fusarium in China[J]. Scientia Agricultura Sinica, 1985, 18(6): 1-6.
[2] Ibrahim F M. A new race of the cotton-wilt Fusarium in the Sudan Gezira[J]. Empire Cotton Growing Review, 1966, 43: 296-299.
[3] Armstrong G M, Armstrong J K. A new race (race 6) of the cottonwilt Fusarium from Brazil[J]. Plant Disease Reporter, 1978, 62: 421-423.
[4] Kim Y, Hutmacher R B, Davis R M. Characterization of California isolates of Fusarium oxysporum f. sp. vasinfectum[J/OL]. Plant Disease, 2005, 89(4): 366-372[2022-10-18]. https://doi.org/10.1094/PD-89-0366.
[5] Holmes E A, Bennett R S, Spurgeon D W, et al. New genotypes of Fusarium oxysporum f. sp. vasinfectum from the southeastern United States[J/OL]. Plant Disease, 2009, 93(12): 1298-1304[2022-10-18]. https://doi.org/10.1094/PDIS-93-12-1298.
[6] Bridge P D, Ismail M, Rutherford M. An assessment of aesculin hydrolysis, vegetative compatibility and DNA polymorphism as criteria for characterizing pathogenic races within Fusarium oxysporum f. sp. vasinfectum[J/OL]. Plant Pathology, 1993, 42(2): 264-269[2022-10-18]. https://doi.org/10.1111/j.1365-3059.1993.tb01499.x.
[7] Davis R M, Colyer P D, Rothrock C S, et al. Fusarium wilt of cotton: population diversity and implications for management[J/OL]. Plant Disease, 2006, 90(6): 692-703[2022-10-18]. https://doi.org/10.1094/PD-90-0692.
[8] 郭慶港, 王培培, 鹿秀云, 等. 棉花枯萎病菌新生理型菌株的分子鉴定[J/OL]. 植物病理学报, 2018, 48(2): 145-153[2022-10-18]. https://doi.org/10.13926/j.cnki.apps.000208.
Guo Qinggang, Wang Peipei, Lu Xiuyun, et al. Molecular identification of the new genotype strains of Fusarium oxysporum f. sp. vasinfectum in China[J/OL]. Acta Phytopathologica Sinica, 2018, 48(2): 145-153[2022-10-28]. https://doi.org/10.13926/j.cnki.apps.000208.
[9] Skovgaard K, Nirenberg H I, ODonnell K, et al. Evolution of Fusarium oxysporum f. sp. vasinfectum races inferred from multigene genealogies[J/OL]. Phytopathology, 2001, 91(12): 1231-1237[2022-10-18]. https://doi.org/10.1094/PHYTO.2001.91.12.1231.
[10] Abd-Elsalam K A, Omar M R, Migheli Q, et al. Genetic characterization of Fusarium oxysporum f. sp. vasinfectum isolates by random amplification of polymorphic DNA (RAPD) and amplified fragment length polymorphism (AFLP)[J]. Journal of Plant Disease and Protection, 2004, 111(6): 534-544.
[11] Assigbetse K B, Fernandez D, Dubois M P, et al. Differentiation of Fusarium oxysporum f. sp. vasinfectum races on cotton by random amplified polymorphic DNA (RAPD) analysis[J/OL]. Phytopathology, 1994, 84: 622-626[2022-10-18]. https://doi.org/10.1094/Phyto-84-622.
[12] Cianchetta A N, Allen T W, Hutmacher R B, et al. Plant pathology and nematology survey of Fusarium oxysporum f. sp. vasinfectum in the United States[J]. The Journal of Cotton Science, 2015, 19(2): 328-336.
[13] 田新莉,趙宗胜,李国英, 等. 新疆棉花枯萎病菌的RAPD分析[J]. 西北农业学报, 2002, 11(4): 4-8.
Tian Xinli, Zhao Zongsheng, Li Guoying, et al. RAPD analysis of Fusarium Oxysporum f. sp. vasinfectum from cotton in Xinjiang[J]. Acta Agriculture Boreali-occidentalis Sinica, 2002, 11(4): 4-8.
[14] 白剑宇. 新疆棉花枯萎病菌生理小种及营养亲和群遗传分化研究[D]. 乌鲁木齐: 新疆农业大学, 2007.
Bai Jianyu. Study on the physiologic race and VCG heredity differentiation of the cotton Fusarium wilt in Xinjiang[D]. Urumqi: Xinjiang Agricultural University, 2007.
[15] 陈文霞. 新疆棉花枯萎病菌遗传多样性研究及其分子检测[D]. 石河子: 石河子大学, 2008.
Chen Wenxia. Studies on genetic diversity and molecular detection of Fusarium oxysporum f. sp. Vasinfectum in Xinjiang[D]. Shihezi: Shihezi University, 2008.
[16] Guo Q G, Li S Z, Lu X Y, et al. Identification of a new genotype of Fusarium oxysporum f. sp. vasinfectum on cotton in China[J/OL]. Plant Disease, 2015, 99(11): 1569-1577[2022-10-
18]. https://doi.org/10.1094/PDIS-12-14-1238-RE.
[17] Tooley P W, Goley E D, Carras M M, et al. Characterization of Claviceps species pathogenic on sorghum by sequence analysis of the β-tubulin gene intron 3 region and EF-1α gene intron 4[J/OL]. Mycologia, 2001, 93(3): 541-551[2022-10-18]. https://doi.org/10.1080/00275514.2001.12063186.
[18] 李红霞, 陆悦健, 王建新, 等. 禾谷镰孢菌β-微管蛋白基因克隆及其与多菌灵抗药性关系的分析[J/OL]. 微生物学报, 2003, 43(4): 424-429[2022-10-18]. https://doi.org/10.13343/j.cnki.wsxb.2003.04.004.
Li Hongxia, Lu Yuejian, Wang Jianxin, et al. Cloning of β-tubulin gene from Gibberella zeae and analysis its relationship with Carbendazim-resistance[J/OL]. Acta Microbiologica Sinica, 2003(4): 424-429[2022-10-18]. https://doi.org/10.13343/j.cnki.wsxb.2003.04.004.
[19] 李双双, 张迎新, 范成明, 等. 单倍型分析技术研究进展[J/OL]. 生物工程学报, 2018, 34(6): 852-861[2022-10-18]. https://doi.org/10.13345/j.cjb.170451.
Li Shuangshuang, Zhang Yingxin, Fan Chengming et al. Advances in haplotype analysis technique[J/OL]. Chinese Journal of Biotechnology, 2018, 34(6): 852-861[2022-10-18]. https://doi.org/10.13345/j.cjb.170451.
[20] Tishkoff S A, Dietzsch E, Speed W, et al. Global patterns of linkage disequilibrium at the CD4 locus and modern human origins[J/OL]. Science, 1996, 271(5254): 1380-1387[2022-10-18]. https://doi.org/10.1126/science.271.5254.1380.
[21] 马晓慧, 李宁, 王大伟, 等. 基于线粒体细胞色素b基因分析海南岛褐家鼠种群遗传多样性[J/OL]. 动物学杂志, 2016, 51(5): 806-816[2022-10-18]. https://doi.org/10.13859/j.cjz.201605010.
Ma Xiaohui, Li Ning, Wang Dawei, et al. Genetic diversity of the Norway rat (Rattus norvegicus) in Hainan Island based on mitochondrial cytochrome b gene[J/OL]. Chinese Journal of Zoology, 2016, 51(5): 806-816[2022-10-18]. https://doi.org/10.13859/j.cjz.201605010.
[22] 李楊, 李河, 周国英, 等. 油茶暹罗刺盘孢菌群体遗传结构分析[J/OL]. 植物保护, 2017, 43(3): 49-54[2022-10-18]. https://doi.org/10.3969/j.issn.0519-1542.2017.03.008.
Li Yang, Li He, Zhou Guoying, et al. Population genetic structure of Colletotrichum siamense on tea-oil trees[J/OL]. Plant Protection, 2017, 43(3): 49-54[2022-10-18]. https://doi.org/10.3969/j.issn.0519-1542.2017.03.008.
[23] Riccioni C, Rubini A, Türkoglu A, et al. Ribosomal DNA polymorphisms reveal genetic structure and a phylogeographic pattern in the Burgundy truffle Tuber aestivum Vittad[J/OL]. Mycologia, 2019, 111(1): 26-39[2022-10-18]. https://doi.org/10.1080/00275514.2018.1543508.
[24] Abd-Elsalam K A, Asran-Amal A, Schnieder F, et al. Molecular detection of Fusarium oxysporum f. sp. vasinfectum in cotton roots by PCR and real-time PCR assay[J]. Journal of Plant Diseases and Protection, 2006, 113(1): 14-19.
[25] Halpern H C, Qi P, Kemerait R C, et al. Genetic diversity and population structure of races of Fusarium oxysporum causing cotton wilt[J/OL]. G3 (Bethesda, Md.), 2020, 10(9): 3261-3269[2022-10-18]. https://doi.org/10.1534/g3.120.401187.
[26] Roger A J, Sandblom O, Doolittle W F, et al. An evaluation of elongation factor 1 alpha as a phylogenetic marker for eukaryotes[J/OL]. Molecular Biology and Evolution, 1999, 16(2): 218-233[2022-10-18]. https://doi.org/10.1093/oxfordjournals.molbev.a026104.
[27] Elias K S, Zamir D, Lichtman-Pleban T, et al. Population structure of Fusarium oxysporum f. sp. lycopersici: RFLP provide genetic evidence that vegetative compatibility group is an indicator of evolutionary origin[J/OL]. Molecular Plant-microbe Interactions, 1993, 6(5): 565-572[2022-10-18]. https://doi.org/10.1094/MPMI-6-565.
猜你喜欢
作文周刊·小学一年级版(2023年36期)2023-09-14
少儿科学周刊·儿童版(2022年10期)2022-06-27
少儿科学周刊·儿童版(2022年10期)2022-06-27
小读者(2020年4期)2020-06-16
学苑创造·A版(2017年9期)2017-09-25
小溪流(画刊)(2017年1期)2017-03-16
小布老虎(2016年8期)2016-12-01
学生天地·小学低年级版(2016年7期)2016-05-14
红领巾·萌芽(2015年7期)2015-09-10
红领巾·萌芽(2015年6期)2015-08-14
