
基于gB基因的IBRV微滴式数字PCR检测方法的建立及初步应用
2023-04-18 03:20高雅欣刘立兵刘岳林王金凤王建昌李睿文河北农业大学动物医学学院河北保定0700石家庄海关技术中心河北石家庄050035
中国兽医学报 2023年3期
高雅欣,刘立兵,刘岳林,王金凤,王建昌*,李睿文* (.河北农业大学 动物医学学院;河北 保定 0700;.石家庄海关技术中心,河北 石家庄 050035)
牛传染性鼻气管炎(infectious bovine rhinotracheitis,IBR)俗称“红鼻病”,是由牛传染性鼻气管炎病毒(infectious bovine rhinotracheitis virus,IBRV)引起的一种急性、热性、接触性传染病。IBRV又称牛疱疹病毒Ⅰ型(bovine herpesvirusⅠ,BHV-1),是一种具有囊膜的双股 DNA 病毒,属于疱疹病毒科(Herpesviridae)、α疱疹病毒亚科(Alphaherpesvirinae)、水痘病毒属(Varicellovirus),是引起牛呼吸系统综合征的最常见的病原之一[1-2]。1956年,MADIN等[3]首次从患病牛中分离出IBRV。1980年,我国首次从新西兰进口种牛中分离得到IBRV,随后在国内部分地区的养牛场均有不同程度的感染和发病[4]。IBRV在临床上常呈隐性感染,终身潜伏在三叉神经节(TG)中,在受到运输、断奶等应激反应后被激活,造成IBRV感染[5]。感染后常引起牛结膜炎、慢性坏死性子宫内膜炎和卵巢炎等疾病,导致妊娠期母牛流产,严重时导致死亡[6-7]。近几年我国的牛群中IBRV的感染率呈上升趋势,给养牛业造成了巨大的经济损失[8-9]。目前尚无特效药物治疗,所以建立精准有效的检测方法,对该病的早期预防和降低病原扩散有重要意义。
微滴式数字 PCR(ddPCR)是一种精准的靶基因定量检测方法,通过对目标靶基因的微滴化处理,经PCR扩增后逐个对每个微滴进行检测,有荧光信号的微滴判读为1,没有荧光信号的微滴判读为0,根据泊松分布原理及阳性微滴的个数与比例即可得出靶基因的拷贝数。该方法不依赖标准曲线实现对靶基因的绝对定量,在灵敏度方面优于实时荧光PCR方法[10-11]。本研究以gB基因为靶基因,建立了IBRV ddPCR检测方法,以期作为临床样品中IBR的精准定量提供一种有效的技术手段。
1 材料与方法
1.1 病毒株和临床样品牛传染性鼻气管炎(IBRV)、腺病毒7型(bovine adenoviruse,BAdV)、牛鼻炎病毒B型(bovine rhinitis B virus,BRBV)、牛病毒性腹泻病毒(bovine viral diarrhea virus,BVDV)、牛冠状病毒(bovine coronavirus,bCoV)、D型流感病毒(inlfuenza D virus,IDV)、牛合胞体病毒(bovine respiratory syncytial virus,BRSV)的DNA或cDNA,均保存于本实验室。119份牛鼻拭子样品,均采自河北省不同地区奶牛场具有呼吸道症状的奶牛。
1.2 主要试剂与仪器ddPCR预混液(不含dUTP)、微滴反应板、微滴发生盖、微滴式数字PCR机油,均购自美国BIO-RAD公司;pMD19-T载体,购自宝生物技术(北京)有限公司;病毒基因组DNA/ RNA提取试剂盒,购自天根生化科技(北京)有限公司;PerfectStar II Probe qPCR superMix(2×),购北京全式金生物技术有限公司。
BIO-RAD QX200 ddPCR微滴生成仪、Droplet Generator Cartidge、QX200TM Droplet Reader和PX1热封仪,购于美国BIO-RAD公司;T-Gradient梯度PCR仪,购于德国Biometra公司;ABI Quant Studio5荧光PCR仪,购于美国ABI公司;NanoDrop2000C,购于美国Thermo Fisher公司。
1.3 引物设计与合成根据GenBank上发布的IBRV基因组序列(NC_001847.1),以gB基因为靶基因,采用DNAMAN软件进行基因序列比对分析,设计合成扩增gB基因全长的PCR引物,同时参考国家标准[12](GB/T 27981-2011)合成ddPCR方法的引物和探针,序列见表1。所有引物、探针均由上海捷瑞生物工程有限公司合成。

表1 引物探针序列
1.4 IBRVgB重组质粒的构建及其鉴定以IBRV基因组DNA为模板,以PCR-gB-F/R为引物进行PCR扩增,将扩增产物纯化、连接和转化到DH5α感受态细胞,挑选阳性克隆由北京擎科生物有限公司测序,应用BLAST软件进行序列分析。使用ND 2000c测定测序正确的阳性质粒浓度,根据公式:拷贝数(copies/μL)= (6.02×1023×质量浓度(mg/L)×10-9)/(碱基数×660),计算重组质粒拷贝数。将重组质粒pMD19-T-gB进行10倍倍比稀释,使其浓度范围为104~10-1copies /μL,于-20℃保存备用。
1.5 ddPCR反应体系和反应条件的优化采用方阵试验对退火温度(55,55.6,56.5,58.1,59.9,61.4,62.4和63.0℃),上下游引物ddPCR-gB-F/R终浓度(400,600,900和1 200 nmol/L),探针ddPCR-gB-P终浓度(200,250,250和500 nmol/L),进行优化。
ddPCR反应体系为20 μL,其中ddPCR Supermix for Probes(no dUTP)10 μL,重组质粒 pMD19-T-gB(1.8×103copies/μL)1 μL,设定上下游引物ddPCR-gB-F/R和ddPCR-gB-P终浓度,补水至20 μL。每个浓度做3次重复。
ddPCR扩增反应条件为95℃预变性10 min; 94℃变性30 s,设定退火温度,延伸1 min,共进行40次循环;98℃酶失活10 min。反应结束后通过微滴读取仪进行读数。根据阳性微滴数、微滴分布状态、微滴荧光信号的强度等确定最佳反应条件。
1.6 特异性试验以IBRV(1.8×103copies/μL)、BAdV-7、BRBV、BVDV、BCoV、IDV、BRSV的DNA或cDNA作为模板,根据优化的反应体系和反应条件进行检测,以分析所建立ddPCR方法的特异性。特异性分析进行3次重复。
1.7 敏感性试验将重组质粒pMD19-T-gB进行10倍倍比稀释,使其浓度分别在1.8×104~1.8×10-1copies/μL之间,并以之作为模板进行ddPCR方法检测,每个浓度梯度重复3次,以分析所建立ddPCR方法的敏感性。
1.8 临床样品检测对采集的119份牛鼻拭子样品,加入PBS缓冲液,充分震荡混匀,采用病毒基因组DNA/RNA提取试剂盒提取病毒核酸,取1 μL为模板,使用本研究建立的ddPCR方法进行IBRV检测,并与GB/T 27981-2011中规定的实时荧光PCR方法检测结果进行比较。
2 结果
2.1 ddPCR方法退火温度优化设定不同的退火温度进行优化。结果显示,当退火温度为56.5℃时,阳性微滴数量均最多,分布更为集中,阴阳性微滴间距最大,微滴荧光信号的最强;3次重复结果一致,因此ddPCR方法的最佳退火温度确定为56.5℃(图1)。确定ddPCR方法的最佳反应条件为:95℃预变性10 min;94℃变性30 s,56.5℃退火延伸1 min,共进行40次循环;98℃酶失活10 min。
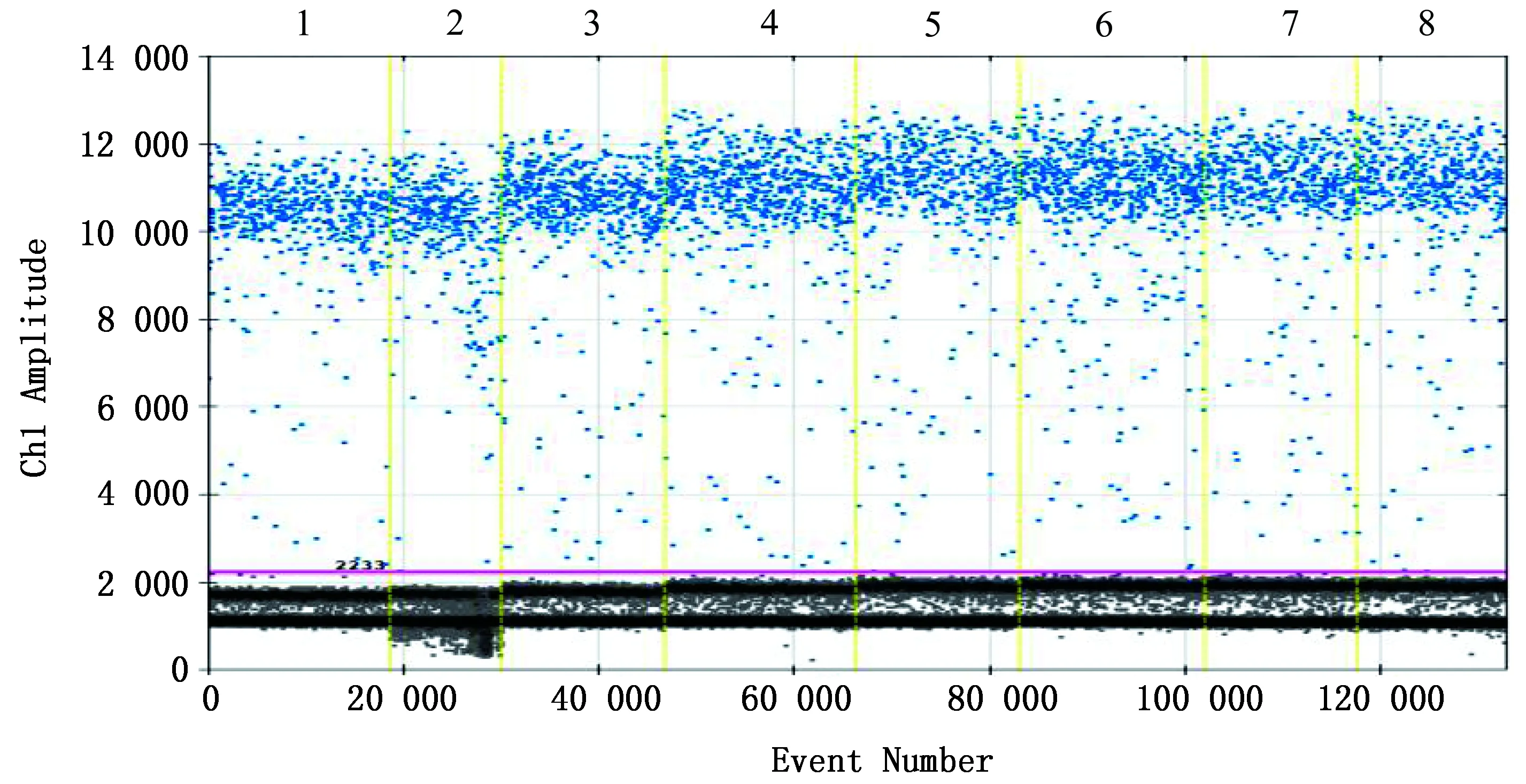
1~8.退火温度分别为63.0,62.4,61.4,59.9,58.1,56.5,55.6和55.0℃
2.2 ddPCR方法引物探针浓度优化在反应体系中设置不同浓度的引物探针进行优化。结果显示,当上下游引物终浓度为900 nmol/L、探针终浓度为250 nmol/L时,阳性微滴数量最多且分布更为集中,阴阳性微滴间距最大,微滴荧光信号的最强,可获得最大扩增效率(图2)。确定ddPCR的最佳的反应体系为:ddPCR Supermix for Probes(No dUTP)10 μL,ddPCR-gB-R/F引物各1.8 μL(终浓度为900 nmol/L),ddPCR-gB-P 0.5 μL(终浓度为250 nmol/L),模板1 μL,水补至20 μL。
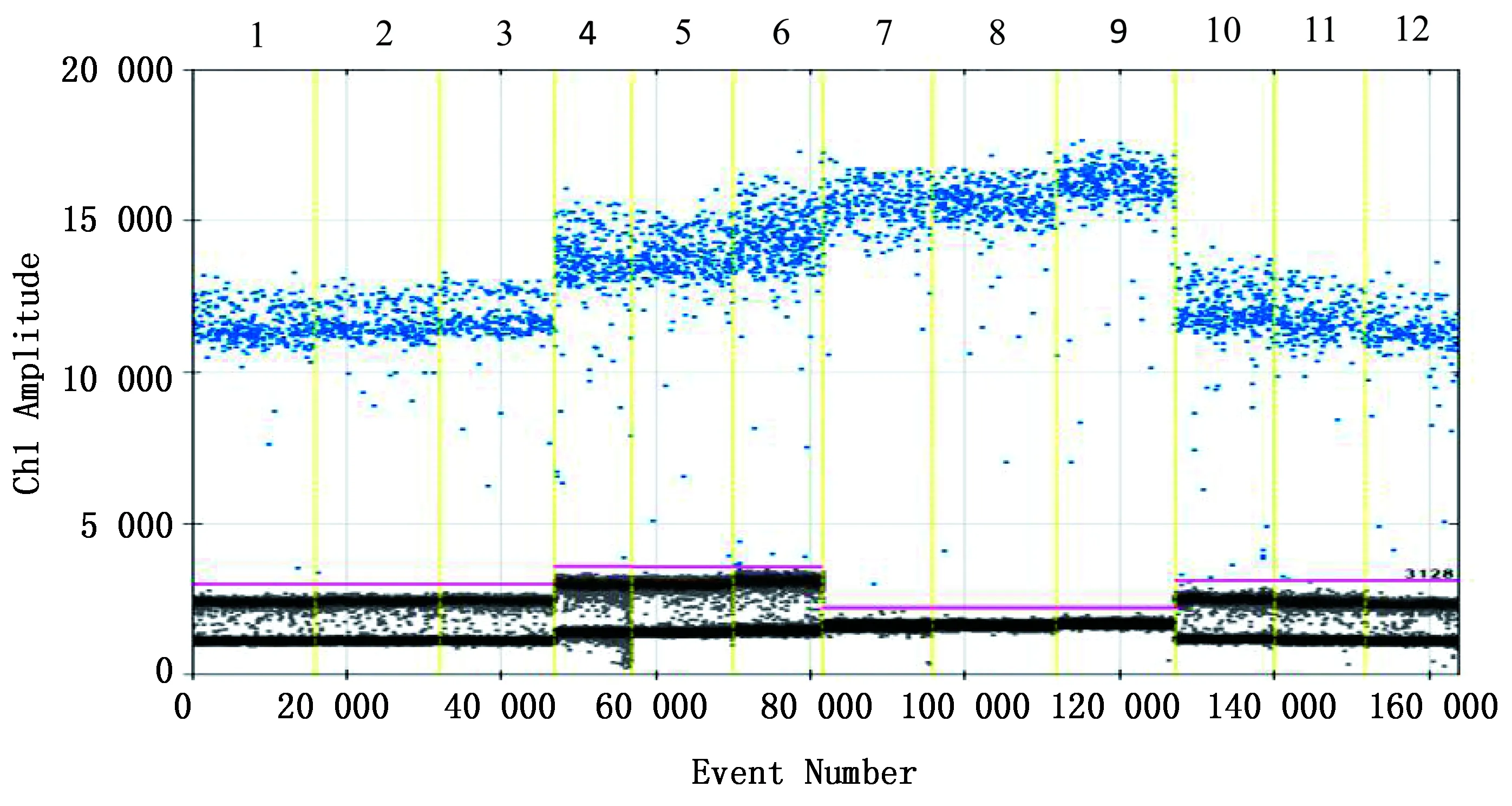
1~3.上下游引物400 nmol/L和探针200 nmol/L; 4~6.上下游引物600 nmol/L和探针250 nmol/L;7~9.上下游引物900 nmol/L和探针250 nmol/L;10~12.上下游引物1 200 nmol/L和探针500 nmol/L
2.3 ddPCR方法特异性试验以BAdV-7、BRBV、BVDV、BCoV、IDV、BRSV基因组DNA/cDNA为模板,进行IBRV ddPCR方法的特异性试验。结果显示,仅IBRV有阳性微滴,其他常见牛呼吸道疫病病原均无阳性微滴,3次重复结果一致,表明所建立的ddPCR方法具有很好的特异性(图3)。

1.IBRV-;2.BAdV-7;3.BRBV;4.BVDV;5.BCoV;6.IDV;7.BRSV;
2.4 ddPCR方法敏感性试验根据上述确定的最佳反应条件和反应体系,以10倍系列稀释的重组质粒pMD19-T-gB为模板,分别进行进行ddPCR方法检测和实时荧光PCR检测。结果显示,建立的 ddPCR方法最低检出限为1.8 copies/μL(图4A),不同浓度3次重复试验结果的变异系数均小于7%(表2),而实时荧光PCR方法最低检出限为1.8×101copies/μL(图4B),表明建立的ddPCR方法灵敏性高、重复性好。

表2 不同浓度的IBRV ddPCR检测结果

A.ddPCR方法的灵敏性试验;B.实时荧光PCR方法的灵敏性试验;1.1.8×104copies/μL;2.1.8×103copies/μL;3.1.8×102copies/μL;4.1.8×101copies/μL;5.1.8×100copies/μL;6.1.8×10-1copies/μL;7.H2O
2.5 临床样品检测采用本研究建立的ddPCR对119份具有呼吸道临床症状的牛鼻拭子样品进行IBRVgB基因检测,并与实时荧光PCR方法进行比较。结果显示,在119份鼻拭子样品中,ddPCR方法检出18份阳性,检出率为15.13%(18/119);实时荧光PCR方法中,检出15份阳性,检出率为12.60%(15/119)(表3)。对3份ddPCR方法检测中为阳性,而实时荧光 PCR方法检测中均为阴性的样品进一步分析,ddPCR方法3次重复试验结果一致,变异系数小于6%(表4)。结果表明,基于IBRVgB基因建立的ddPCR方法敏感性高于实时荧光 PCR方法,且检测结果稳定、可靠,能够实现样品中病毒载量的定量分析,更适合病毒含量低的鼻拭子样本检测。
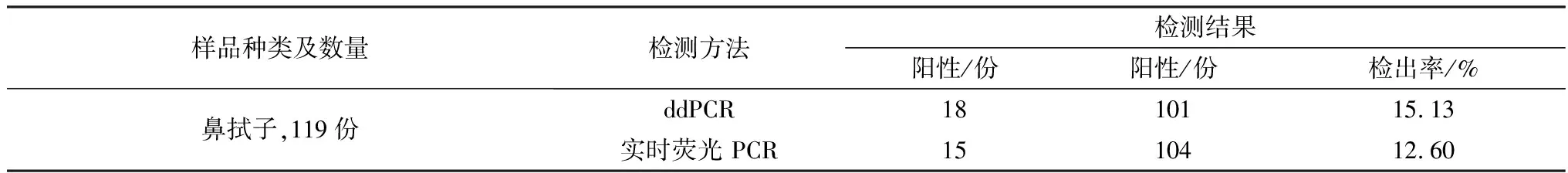
表3 IBRV微滴式数字PCR和实时荧光PCR的临床样品检测结果

表4 ddPCR检测重复性试验
3 讨论
IBRV是引起牛呼吸道综合征的重要病原体之一,在世界范围内广泛存在,对牛养殖业造成了较大的经济损失。其具有持续感染和隐性感染的特点,目前针对该病毒无特效药物,不能完全清除。因此,早期快速诊断在 IBR 防治中起着至关重要的作用。国内外学者针对IBRV建立了PCR方法、实时荧光PCR方法[13-14]、环介导等温扩增(LAMP)方法[15]和重组酶聚合酶扩增(RPA)方法[16]。其中,荧光定量PCR方法在实验室检测中应用最为广泛,与PCR方法相比,检测效率较高,在特异性和灵敏性方面有很大优势,但针对鼻拭子等病毒含量较低的样品,阳性检出率很低,很难实现IBR早期的精准检测。基于LAMP技术建立的IBRV等温扩增检测技术,具有成本低、操作简便、能够应用于现场的快速检测等优点,但该方法引物设计较为复杂,而且假阳性率较高。RPA技术做为现场诊断的一种等温扩增技术,具有检测速度快、灵敏度高、特异性好等特点,适合于临床样品现场的快速检测,但在精准定量检测方面存在一定的局限性。微滴式数字PCR方法已经广泛应用于病原检测领域,检出限可以达到单个拷贝,尤其适用于对低拷贝、低含量病原的精准检测,可以有效弥补其他方法的不足[17]。
本研究基于gB基因建立了IBRV的ddPCR方法,通过对ddPCR退火温度、引物探针浓度的优化,确定了最佳的反应条件和反应体系;所建立的ddPCR对引起牛常见呼吸道疫病病原均无交叉反应,具有较强的特异性;敏感性是荧光定量PCR方法敏感性的10倍,检出限可达到1.8 copies/μL。在临床样品检测中,ddPCR方法检出18份阳性,实时荧光 PCR方法检出15份阳性,其中3份样品ddPCR方法检测的平均微滴数分别为7.23,7.90和7.90 copies/μL,而实时荧光 PCR方法检测均为阴性。对于IBRV含量较低的样品,尤其是处于实时荧光 PCR方法检测中的可疑或无法判定的样品,ddPCR方法可以实现对样品中靶基因的精准检测,有效分析鼻拭子样品中IBRV的含量,从而实现对IBRV感染的早期监测和精准预防。但ddPCR方法有一定的不足,该方法需要更为昂贵的仪器设备、装备良好的实验室环境,以及对技术人员的要求更高,试剂耗材成本更高,广泛推广难度较大,无法实现养殖场的现场诊断。
本研究建立的IBRV ddPCR检测方法,具有特异性好、灵敏度高,可以实现对临床样品中IBRV的精准定量检测,为牛场中IBRV感染的监测提供了一种新的有效的技术手段。