
第二类医用电气设备抽检问题分析与监管建议
2023-03-30 07:00王志远黄珊珊
中国药业 2023年6期
周 冬,齐 健,吴 璠,王志远,黄珊珊
(安徽省药品审评查验中心,安徽 合肥 230051)
有源医疗器械是指依靠电能或其他能源,而不是直接由人体或重力产生的能量,发挥其功能的医疗器械[1]。医用电气设备是有源医疗器械的重要组成部分,近年来国家医疗器械质量监督抽检(以下简称“国抽”)结果显示,医用电气设备不符合标准规定的检出比例较高[2-4]。自2014 年版《医疗器械监督管理条例》实施以来,药品监督管理部门不断加强医疗器械质量抽检力度。2021 年6 月新修订《医疗器械监督管理条例》正式实施,从制度层面进一步推动行业创新,更好地满足了人民群众对高质量医疗器械的期待。“国抽”是加强医疗器械质量监管、维护公众用械安全的重要手段,相关检查结果在国家药品监督管理局网站医疗器械板块定期公示。本研究中基于2014 年至2021 年公布的“国抽”通告中涉及第二类医用电气设备产品检验结果,通过分析不符合标准规定项目情况,探讨第二类有源医疗器械质量风险和存在问题,并从上市前注册和上市后监管等多角度提出监管对策和建议,以期督促第二类有源医疗器械生产企业严控产品质量安全,不断提升医疗器械质量安全保障水平。现报道如下。
1 资料与方法
根据国家药监局网站医疗器械板块公布的2014年1月至2021年12月“国抽”结果通告,选取第二类有源医疗器械中的医用电气设备产品不符合标准规定的项目及数据,按2017版《医疗器械分类目录》进行分类。采用WPS 2012软件对不符合标准规定项目类型进行统计和分析。
2 结果与分析
2.1 抽检结果
总体不合规情况:其发现不符合标准规定项目581项,涉及产品428 批,共13 大类。产品数量(批次)排名前3 的产品类别为物理治疗器械,医用诊察和监护器械,以及注输、护理和防护器械,详见表1。不合规项目类型包括电气安全(252 项,43.37%),标识、标记或随机文件(168项,28.91%),产品性能(154项,26.51%)和不能正常使用(7项,1.20%)。
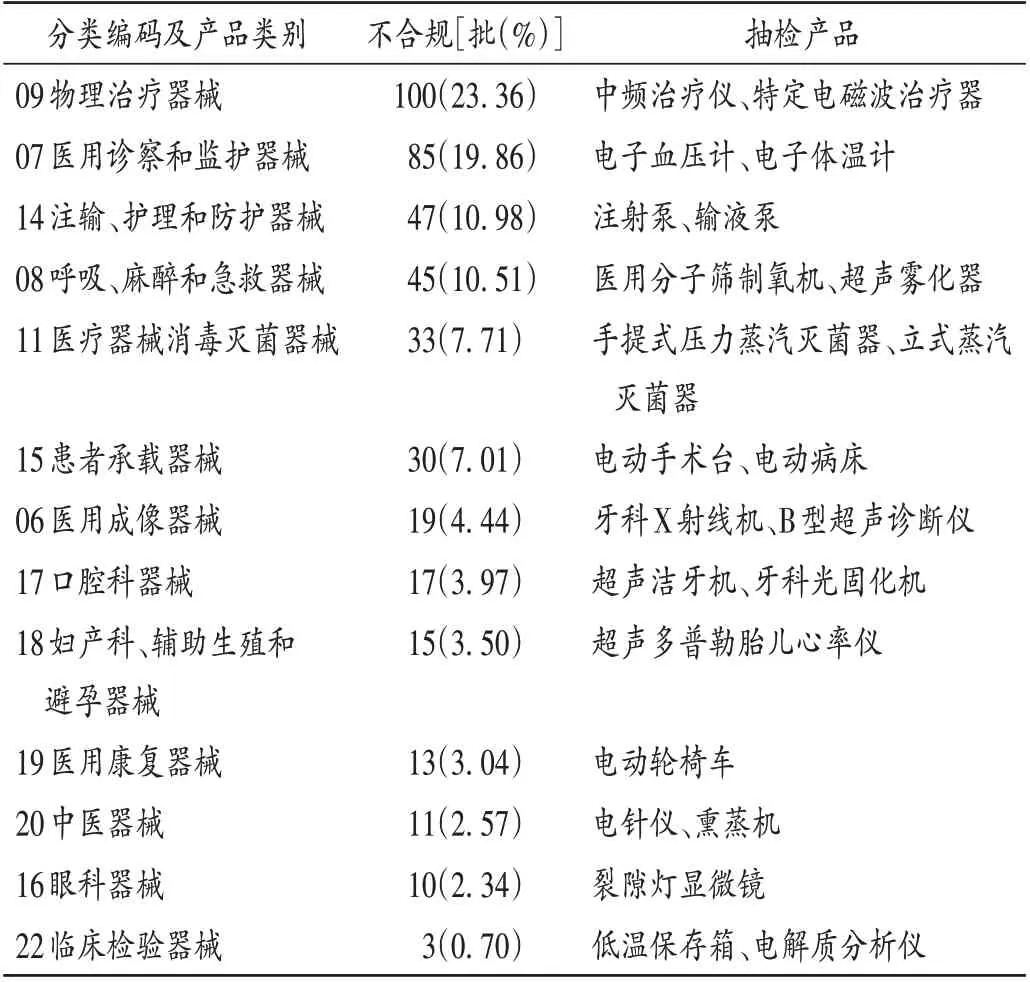
表1 2014年至2021年“国抽”医用电气设备产品不合规情况(n=428)Tab.1 Fields involved in the classification of medical electrical equipment products in national quality supervision and sampling inspection from 2014 to 2021(n=428)
不符合标准规定项目类型:按产品类别对不符合标准规定的主要项目类型分别统计,结果详见图1。总体来看,“国抽”中医用诊察和监护器械以及物理治疗器械不合规项目数整体偏多。
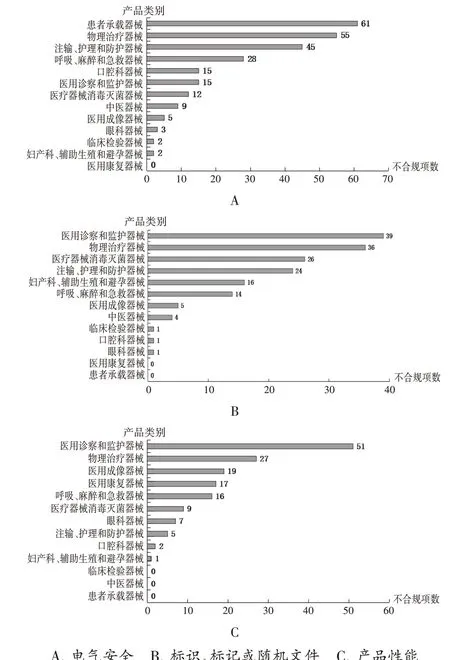
图1 不合规医用电气设备产品类别分布A.Electrical safety B.Identification,marking or random files C.Product performanceFig.1 Category distribution of medical electrical equipment products
2.2 问题分析
2.2.1 不合规情况涉及问题
标识、标记或随机文件:包括产品的防电击类型、程度,电源参数,工作方式等安全特征信息,需在随机文件中详细写明,特别是外部标记应是“永久贴牢的”和“清楚易认的”。经分析,在不符合标准规定的标识、标记或随机文件中,外部标记占30.95%,标识要求占10.12%,随机文件占13.10%。存在问题主要有标识缺失、标识不明确或错误、标记的耐久性不够等。
电气安全:其为有源医疗器械产品的关键性能指标,检验依据主要包括GB 9706.1 —2007 和GB 4793.1 —2007 标准,检验项目包括正常工作下连续漏电流和患者辅助电流,输入功率,保护接地,电源供电的中断,与供电网的分断,熔断器,外壳和防护罩等。在不符合标准规定的电气安全类别中,输入功率占14.68%,运动部件占11.11%。存在问题主要有元器件材料、电流电压电阻功率、结构安全、意外提示报警及保护等。
产品性能:性能指标是指可进行客观判定的成品的功能性、安全性指标,产品性能属功能性指标,存在的问题主要有控制器件的准确性,功能性指标(如氧浓度、最大速率等)的可及性和可靠性,操作部件的设计等问题。
2.2.2 按注册技术审查指导原则分析
注册技术审查指导原则作为指导性文件,适用于企业准备其产品注册申报资料和技术审评人员对产品上市前申报材料的审查,能指导和规范医疗器械产品的技术审评工作[5]。按产品类别进行统计,不符合标准规定产品共涉及35 个一级产品类别和54 个二级产品类别,其中28 个(80%)一级产品类别的产品已出台相关注册技术审查指导原则,表明监管部门已从上市前注册管理环节对重点抽检产品统一技术审评尺度,对产品的安全性和有效性审查提出了明确标准。但仍有部分使用量大的医用电气设备产品尚未制订相关指导原则,如特定电磁波治疗器、电针仪等。详见表2。
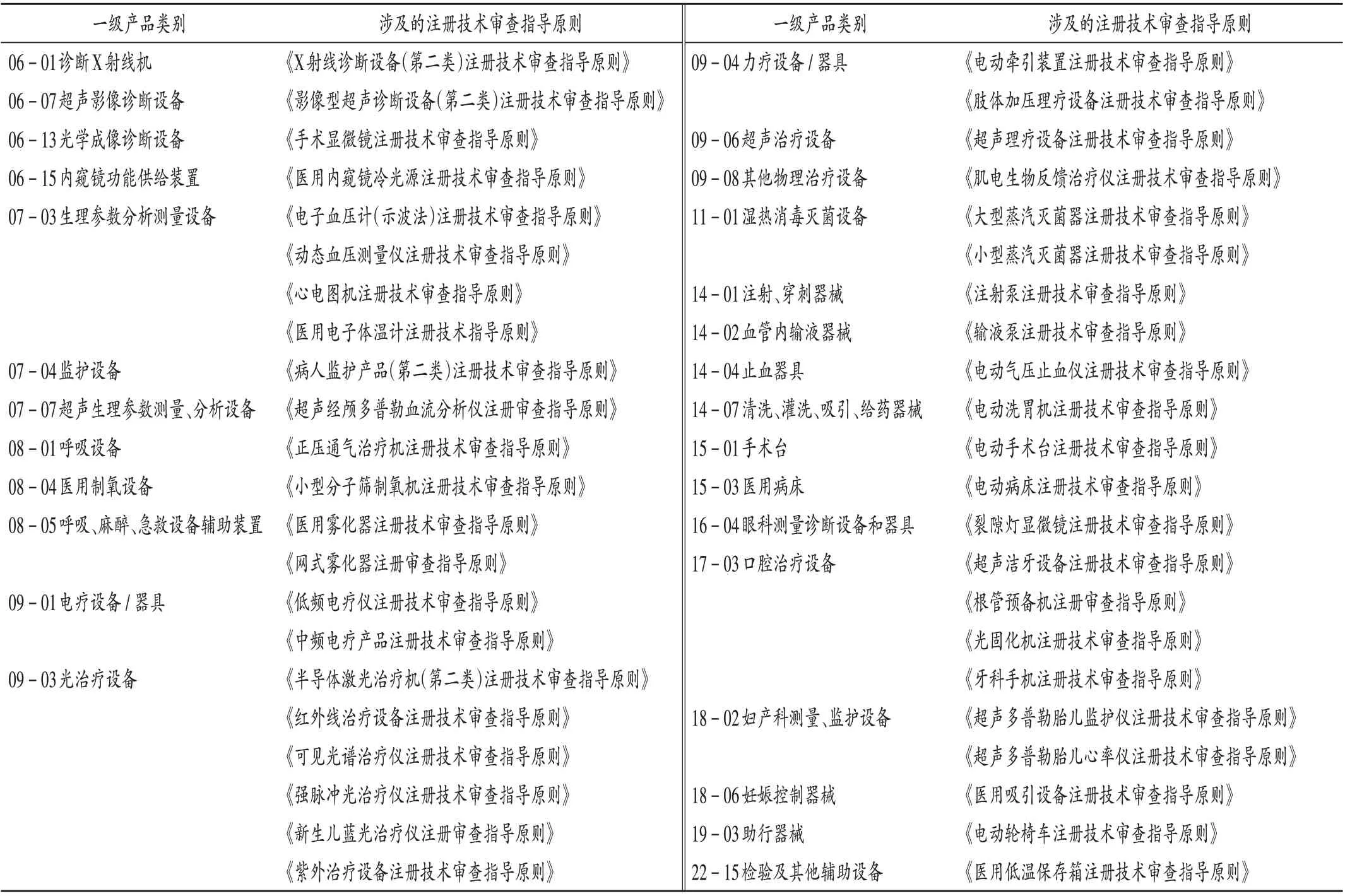
表2 不合规产品涉及注册技术审查指导原则分布Tab.2 Distribution of non-conforming products involved in the technical review guidelines for registration
2.2.3 按生产环节风险清单和检查要点分析
医疗器械生产环节风险清单和检查要点作为实施监督检查的重要指南,能增强监管人员的风险识别能力,提升生产企业的风险防控水平。原国家食品药品监督管理总局自2016 年起共印发了46 种医疗器械生产环节风险清单和检查要点[6-7],但90%以上的产品为第三类医疗器械,仅涉及部分第二类无源医疗器械(如定制式义齿、医用防护服、医用防护口罩)。依据现行分类规则,呼吸机和电动输液泵产品分别有界定为第二类医疗器械的无创呼吸机(用于非生命支持)和输液泵(不包含贮液装置和输液管路)。经分析,不符合标准规定产品类别涉及上述2种医用电气设备,分别为08-01呼吸设备和14-02 血管内输液器械。提示监管人员可基于产品生产工艺的相似程度,视情况采用风险清单和检查要点中相关内容供上市后监督检查参考。
2.2.4 第二类医用电气设备质量风险分析
企业对“国抽”不符合标准规定情况的关注度不够:2020 年颁布的《医疗器械质量抽查检验管理办法》(国药监械管〔2020〕9 号)为规范医疗器械质量抽查检验工作进一步明确了工作思路。针对第二类有源医疗器械,“国抽”通常将临床用量大、使用人群和使用范围广的、不良事件监测提示可能存在质量问题的产品作为重点抽检对象。但多数企业对“国抽”结果关注度较低,缺乏风险意识,对于同品种公布的不符合标准规定的问题、因质量不合格可能导致的不良事件及产品召回等警示信息未能作为可预见风险在纳入设计开发环节进行风险管理,无法识别风险成因和开展风险防控,可能造成企业自身产品在各级抽检中出现类似不符合标准规定的问题。
企业内部质量管理不到位,未严格依法依规组织生产:“国抽”结果显示,多数问题集中在未有效贯彻产品性能指标中的“安全”要求。企业在上市前注册检验时产品技术要求虽能严格按GB 9706.1 —2007 和GB 4793.1 —2007 等强制性标准要求制订“安全”要求,并在产品说明书中对设备上的图形、符号进行解释说明,确保随机文件(使用说明书、技术说明书)告知用户信息的完整性。但由于实际生产过程中质量控制不到位,未严格贯彻强制性标准和(或)产品技术要求,导致产品性能指标特别是“安全”要求未能符合标准,造成医用电气设备电气安全方面存在风险。如有的设备输出热能时无标示明显的指示器,也未按要求安装黄色指示灯指示设备的输出状态,可能使患者在不知道设备已工作的情况下接触设备的辐射器而造成意外烫伤。
企业对产品说明书和标签重要性认识不到位:基于风险管理原则,标识、标记和随机文件通过警告和说明的方式降低医疗器械剩余风险,成为保障医疗器械安全和有效的最后一道风险防控屏障[8]。《医疗器械说明书和标签管理规定》(原国家食品药品监督管理局令第6 号)明确规定了说明书和标签应包括的内容。医用电气设备通用安全标准GB 9706 系列、GB 4793 系列及YY/ T 0466.1—2009 等系列标准明确了医疗器械标签、标记和提供信息的符号的通用要求。注册技术审查指导原则也针对产品特性,并结合各类产品适用标准的通用要求和专用要求,在法规基础上进一步细化了产品说明书和标签的技术审查要求。但企业在实际规模化生产过程中对说明书和标签与标准或产品技术要求的符合性认识不到位,可能存在随意变更上市前注册检验时已依据法规、标准和指导原则要求核准的标识、标记和随机文件内容等情况,造成产品上市后不符合标准规定。以医用吸引设备产品为例,企业未遵循标准和指导原则要求,对于用于野外和/或运输中的电动吸引设备,且不符合YY 0636.1—2008标准中53.1条的要求时,企业未在其外箱标签上标明“不适宜XX ℃以下(或以上)使用”,导致与实际核准的标签信息不一致。
企业对产品设计开发环节管控不到位:已上市产品是经过法定核准的,产品应确保设计定型、结构组成明确、生产工艺稳定,但部分企业缺乏法规意识,实际投产时未按核准的产品结构组成进行生产。同时对技术参数理解不充分,不了解医用电气设备的要求,采购不符合医用电气产品要求的原材料,对于因产品设计、原材料(电子元器件、电源等)等发生可能影响产品质量的实质性变化,也未能开展上市后研究,并及时通过设计验证(检验)和注册等方式确认产品的安全性、有效性和质量可控性,造成产品不符合国家、行业标准和经注册核准的产品技术要求。
企业主体责任还需进一步落实,风险意识亟待提升:上市前注册检验报告仅对送检样品的质量负责。检验过程中出现的技术改进和整改缺陷应纳入设计更改。技术审评过程中重新核准产品型号、修订产品说明书也应纳入设计更改。所有设计更改完成后需及时更新设计输出文件,包括元器件清单、使用说明书、技术说明书、标签和生产工艺规程等。但企业研发人员、生产管理和质量控制人员在设计评审时未能充分沟通确认设计更改完成后的设计输出文件内容,造成实际生产时未按更改后的程序文件和作业指导书要求规范生产,产品上市后经抽检才发现不符合标准规定情况。如有的产品设备上的铭牌标示的产品型号与注册证不符,有的产品说明书上设备型号的操作面板与实际设备不相符。
3 对策与建议
3.1 加大宣传贯彻力度
《医疗器械注册与备案管理办法》(国家市场监管总局令第47 号)规定,医疗器械注册应遵守相关法律、法规、规章、强制性标准,以证明产品的安全、有效和质量可控。对于有源医疗器械,产品标准既多且杂,企业在执行国家、行业标准时要透彻理解标准含义,重视包装、说明书信息,确保信息的一致性,但不同企业对标准理解存在偏差,可能导致标准在执行过程中贯彻不到位。建议标准管理部门、相关标准化技术委员会和检验检测机构对质量抽检过程中存在的争议问题、共性问题和探索性研究问题,主动面向企业开展标准宣传与贯彻。特别对于即将实施的新版医用电气设备通用安全标准GB 9706.1—2020 及其引发的标准升级变化,应多渠道进行标准宣贯,提供检验检测机构和企业面对面沟通交流机会,督促企业加紧熟悉掌握新标准的内容,提高对标准的理解水平,提前按照新标准的要求提前开展产品设计研发和标准升级等相关工作。
3.2 严格上市前注册管理
技术审评和体系核查作为上市前注册管理的重要环节,相辅相成,密不可分[9-10]。从“国抽”结果分析,不符合标准规定问题涉及电气安全,标识、标记和随机文件,产品性能,建议技术审评环节加大对第二类有源医疗器械电气系统安全性、化学/物理性能研究注册资料的审查,严格审核产品采标方法、采标依据和研究数据;按法规、标准和指导原则要求细化说明书标签内容审查要求,重视推荐性行业标准中的安全性指标的把控,特别要关注通用标准之外专用标准的要求。对于尚未出台指导原则的产品,审评机构可通过制订技术审评要点统一技术审评尺度,规范技术审查要求,避免因注册申请人疏忽和技术审评不细致而引发的上市后质量安全问题。针对体系核查,建议基于“国抽”发现问题,制订个体化核查方案,明确核查方案中重点关注的质量风险,结合国家药品监督管理局新出台的《医疗器械注册自检管理规定》要求,对“国抽”中出现过的不符合标准规定的电气安全性能指标,如连续漏电流和患者辅助电流、电介质强度等,专门选派具备医疗器械检验背景的检查员对企业质检人员的成品检验能力进行现场实操考查,确保出厂检验质量。建议对设计开发环节开展核查时,重点核实注册送检样机的设备或设备部件的外部标记、警告标记等是否与检验报告中图片描述一致,如果注册送检过程中发生了设计更改,需要核实是否及时识别更改内容并更新设计输出要求,从严从细核实样品真实性和设计开发过程的合规性。
3.3 加大上市后监管力度
第二类医疗器械具有中度风险,需要严格控制管理,相较于第三类医疗器械,其产品整体技术含量较低,入行门槛不高。从“国抽”结果来看,一些入行时间较短的第二类医疗器械生产企业,在产品设计开发过程中可能对强制性标准理解不到位,导致生产出不合格产品。同时企业未制定并严格执行出厂检验规程,导致成品质量不可控。此外医疗器械产业发展推动产品不断升级,新产品及换代产品的技术要求、说明书未能在设计输出时及时更新,造成生产管理不可控,也可能导致产品质量问题。建议定期汇总国家级和省级第二类有源医疗器械质量抽检的结果,按重点品种进行质量分析和评估,对发现存在质量问题的企业开展约谈,针对出现不符合标准频率较高、涉及面较广的检验项目,持续对企业开展专题培训,加深企业对标准的理解,确保产品质量。同时建议加强上市后监管的靶向性和针对性[11],可参照国家已出台的风险清单和检查要点,结合质量抽检发现问题和本地区产品特点,从质量风险角度制定省级第二类有源医疗器械重点品种现场检查风险清单和检查要点,加强上市后监督检查和省级监督抽检力度,高度重视因质量问题引发的产品不良事件,特别关注设备外部标记问题、安全规范检测能力等要求,全面提升监督检查质量,督促企业持续提升保障产品质量合规的能力。
3.4 提前介入并靠前服务
随着医疗卫生支出和健康意识增强,医疗器械产业发展正进入高速发展期,“国抽”产品类别显示,物理治疗类、医用诊察和监护类第二类有源医疗器械(涉及中频治疗仪、医用分子筛制氧机、医用雾化器、电子血压计等)生产企业数量多,因此,推动更多优质、合格的医疗器械产品上市,高质量服务医药卫生体制改革及生命健康产业发展迫在眉睫。建议省级检验检测、审评审批等部门源头施策,提前介入,靠前服务,畅通渠道,对医疗器械生产企业做好上市许可前的政策指导和技术帮扶,普及新出台、新修订的法律法规、检验标准、审评指导原则和检查指南等知识;突出研审联动,特别是对于创新和优先审批的医疗器械,积极对接产业集中地区主管部门,上门集中开展专题咨询,面对面答疑解惑,提升企业产品、产品技术要求、产品说明书和标签质量,帮助企业加深对法规、标准和指导原则的认识,真正从源头控制风险,助力医疗器械产业高质量发展。
3.5 加强质量安全风险会商
医疗器械属于特殊商品,产品质量安全关乎生命安全。建议落实医疗器械全生命周期管理理念,完善省级医疗器械检验检测、审评审批、监督检查,不良事件检测、投诉举报、案件查处、舆情监测等多部门质量安全风险定期会商制度。基于问题治理导向,对于涉及多次抽检不合格产品的企业、抽检不合格率高或不良事件监测预警[12]的产品,应加强质量抽验数据分析和利用,多维度分析研判第二类有源医疗器械风险点,理清企业风险、产品风险,聚焦重点产品、问题产品,实施风险清单和质量风险闭环管理,对上市后出现质量问题的企业和产品加强日常监管力度,同时充分利用飞行检查、有因检查和监督抽验[13]等手段及时防控质量安全风险,压实企业主体责任意识,倒逼企业不断提升履责能力和管理水平,严防严控医疗器械质量安全,不断提升医疗器械质量安全保障水平。
猜你喜欢
中国化肥信息(2022年3期)2023-01-05
石材(2020年11期)2021-01-08
中学生数理化·高一版(2020年6期)2020-12-17
中学生数理化(高中版.高二数学)(2020年6期)2020-12-04
小猕猴智力画刊(2020年9期)2020-09-04
医疗装备(2020年10期)2020-06-13
质量安全与检验检测(2019年3期)2019-07-31
质量安全与检验检测(2018年6期)2018-12-28
电子制作(2018年22期)2018-12-23
消费导刊(2018年10期)2018-08-20
