
The anodically polarized Mg surface products and accelerated hydrogen evolution
2023-03-24 09:24JufengHungGungLingSongYixingZhuDjingZhengZimingWng
Journal of Magnesium and Alloys 2023年1期
Jufeng Hung ,Gung-Ling Song,b,c,∗ ,Yixing Zhu ,Djing Zheng ,Ziming Wng
a Center for Marine Materials Corrosion and Protection,College of Materials,Xiamen University,People’s Republic of China
b State Key Laboratory of Physical Chemistry of Solid Surfaces,Xiamen University,Xiamen 361005,People’s Republic of China
c School of Engineering,The University of Queensland,St.Lucia,Qld 4072,Australia
Abstract To clarify the anodic dissolution mechanism of Mg,the hydrogen evolution from pure Mg in acidic solutions under galvanostatic conditions were systematically measured.With increasing anodic current density,the cathodic hydrogen evolution rate decreased,and the anodic hydrogen evolution became faster while some surface area on the Mg was becoming dark under anodic polarization.Based on the surface analysis results and the generally accepted basic electrochemical equations,the evolution kinetics of hydrogen from Mg was deduced,and the most possible surface intermediate active species that could facilitate the anodic Mg dissolution and anodic hydrogen evolution were proposed.This paper further develops the model of incomplete fil Mg+ dissolution,explains many reported experimental phenomena,and clarifie misunderstandings of current mechanism.
Keyword: Magnesium;Corrosion;Hydrogen evolution;Negative difference effect.
1.Introduction
Magnesium(Mg)alloys have various potential applications not only because of their low density,high specifi strength and easy recyclability [1–4],but also due to its unique chemistry.For example,due to the negative open-circuit potential(OCP),non-toxicity and good biocompatibility of Mg,Mg alloys have been made into sacrificia anodes [5] and degradable implants [6,7].Recently,Mg was even developed into a smart corrosivity detector and intelligent corrosion protector for reinforce concrete [8].
In fact,all the traditional and innovative applications of Mg alloys are critically associated with their polarization behaviors,which are fundamentally governed by their electrochemical mechanism.Currently,the electrochemical corrosion of Mg has been intensively studied [9,10],and is generally described as a reaction of Mg with water:
in which hydrogen evolution reaction (HER) is the major cathodic reaction (while oxygen reduction is usually believed to be negligible [11,12]),and Mg dissolution is the main anodic reaction.The well-known negative difference effect (NDE) or anodic hydrogen evolution (i.e.,the rate of HER increases as the polarization current density or potential becomes more positive,which is contradictory to a normal cathodic hydrogen evolution (CHE) process) is normally involved in the anodic dissolution of Mg [12–17].
To explain the hydrogen evolution accelerated by anodic polarization,various models have been proposed,including the partially protective surface film uni-positive Mg+ion dissolution,magnesium hydride (MgH2),particle undermining,incomplete fil univalent Mg+dissolution mechanism,and cathodic catalytic theories [14,15,18–20].Nowadays,the two main models are relatively widely referenced:
1) the mechanism of incomplete fil Mg+dissolution[12,14,17],and
2) the mechanism of enhanced catalytic activity [20,21].
The original univalent Mg+ion theory [22] suggests that intermediate univalent Mg+ions can be produced during Mg dissolution,which may stay in water for a period of time for detection.This theory [12,14,23] was further developed that the univalent Mg+ions could only survive next to the substrate metallic Mg at the breaks (including the pores) of the MgO or Mg(OH)2surface fil or product layer,which could quickly react with H2O to turn into Mg2+.It should be appreciably acknowledged that a significan step forward in this model was made by Barilet al.[24] who believes that the univalent Mg+ion is adsorbed on the Mg surface before further reaction with water or being electrochemically anodized.The existence of Mg+in the solution extended to the Mg surface.Obviously,it would be difficul to detect the univalent Mg+ions in solution far away from the substrate Mg surface,or by Raman spectroscopy that cannot reach inside the pores of the surface fil and does not have a reliable standard univalent Mg+ion peak as reference for comparison to verify the presences of Mg+.
According to the model of incomplete fil Mg+dissolution,in the fil broken area,the substrate Mg is electrochemically dissolved into univalent Mg+ions [25,26]:
which is the rate-determining step of the whole anodic process under ambient conditions.Immediately,a fraction (k) of the univalent Mg+ions is further anodized to Mg2+ions through the electrochemical reaction:
Meanwhile,the remaining univalent Mg+ions (i.e.,(1-k)of the univalent Mg+ions) can actively react with water via:
or equivalently
releasing hydrogen gas.This hydrogen generation process is a result of the anodic dissolution (2) of Mg,termed as anodic hydrogen evolution (AHE).With the above equations(2) through (4a,4b or 4c),the overall anodic process can be expressed as:
while reactions (2),(3)and(4a,4b or 4c)(or reaction(5))are taking place at the fil breaks or in the pores of the surface product layer,where the dark corroding regions or the areas appeared full of non-protective dark corrosion products.The dark corrosion product/area on a Mg alloy surface under anodic polarization condition was firs reported by Song[12,27].The normal cathodic hydrogen reaction can also occur there(i.e.,on the metallic Mg surface at the fil breaks or in the surface layer pores,and some limited hydrogen reaction on the fil covered areas as well):
With increasing anodic current or potential,more univalent Mg+ions will be generated,and more hydrogen gasses produced.Although the model of incomplete fil Mg+dissolution can explain almost all the reported experimental phenomena of Mg corrosion,the critical intermediate active univalent Mg+ion that can be rapidly oxidized into Mg2+by water next to the substrate Mg surface so far has not been experimentally detected.
The hypothesis of enhanced catalytic activity [20,28,29]assumes that there are some catalytic active sites on the Mg surface during anodic dissolution,which can enhance the cathodic hydrogen evolution rate.Apart from the critical catalyst that is not believed to be univalent Mg+ion,the differences of this model from the model of incomplete fil Mg+dissolution basically include:
1) The elementary anodic dissolution of Mg is a 2-electron reaction:and no intermediate specie like the univalent Mg+ion is involved in the reaction.
2) The hydrogen evolution that can be accelerated by increasing polarization potential or current density under anodic polarization is still a normal cathodic process from anodic dissolution region (6a or 6b).
To validate this hypothesis,many studies have been carried out to identify the cathodic catalyst [30,31].After the contribution of the noble impurities [32–34] to the cathodic hydrogen activity has proved to be negligible based on many experimental results [11,35],recent research [35,36] has been focused on the dark corrosion products formed on Mg surface during anodic polarization.However,what the critical catalyst is so far has not been theoretically asserted,nor experimentally detected [35,37].It has even been proposed that the hydrogen evolution may result from complicated electrochemical reactions involving multi-electrons and intermediates like Mg∗H and Mg∗OH [21].However,the possibility of a multi-electron elementary reaction in electrochemistry is very low,and the intermediates Mg∗H and Mg∗OH were not catalysts.In fact,these intermediates proposed by Yuwonoet al.[21] consist of a uni-valence positive magnesium ion in nature,which actually supported the incomplete fil Mg+mechanism.
Different fundamental understandings of the corrosion mechanism of Mg may lead to different protection/prevention strategies for Mg alloys in engineering.For example,a correct identificatio of the surface intermediate species responsible for the accelerated anodic hydrogen evolution and Mg dissolution will concern the selection of suitable service environments and inhibitors for Mg alloys,as the environmental ions may directly interact with the surface intermediate species[38],and thus significantl affect the corrosion resistance of the alloys and the inhibition efficien y of the inhibitors[39]in industry.Therefore,from a practical and scientifi point of view,it is of great significanc to clarify the electrochemical corrosion mechanism in order to approach an unambiguous mechanistic understanding for Mg in corrosion science.
Recently,a comprehensive comparison between the models of the enhanced catalytic activity and the incomplete fil Mg+dissolution was made to answer the question “what activates the Mg surface” [16].Although many key points have been presented in the review,they need detailed theoretical analyses and systematic experimental results to support.These theoretical and experimental results are now provided in this study.This paper aims to deepen current understanding of the corrosion mechanism of Mg based on an objective analysis of the anodically accelerated hydrogen evolution and Mg dissolution,as well as the dark corrosion products or corroding areas,in an acidic solution.Since under such an acidic condition,a surface fil or deposited corrosion product layer normally becomes less stable or even cannot survive,it is expected that the this study will provide a new dimension to unveil the possible immediate active species on Mg and gain a profound insight into the anodically accelerated hydrogen evolution and Mg dissolution.
2.Experimental
2.1.Material and solutions
Mg specimens(15 mm×15 mm×15 mm)were cut from a Mg ingot (0.1066 wt.% Al,0.0046 wt.% Zn,0.0022 wt.%Fe,<0.0006 wt.% Cu,<0.0001 wt.% Ni,<0.0001 wt.% Zr and Mg balance),measured by Icap7200 ICP-OES.The specimen was mounted in epoxy resin after a copper wire was electrically connected to its back and made into an electrode.The surface not sealed by the epoxy was mechanically abraded using SiC paper up to 2000 grit,rinsed with high purity water and ethanol,and then immediately dried in a stream of warm air.
The electrolyte solutions,0.2 M hydrochloric acid (HCl),0.1 M HCl,0.01 M HCl and 0.05 M sulfuric acid (H2SO4),were prepared using analytical grade reagents and high purity water (resistivity~18.2 MΩ•cm).
2.2.Electrochemical measurements
A Gamry Reference 600TMpotentiostat and Gamry Frame workTMsoftware package were used.A three-electrode electrolytic cell with a platinum foil counter electrode and a KClsaturated Ag/AgCl reference electrode or Hg/Hg2SO4reference electrode containing the HCl solutions and the 0.05 M H2SO4solution was used in the electrochemical measurements.All the measured potentials by this reference electrode were converted to those relative to the KCl-saturated Ag/AgCl.
Under anodic galvanostatic polarization conditions,hydrogen gas collection was carried out.The Mg working electrode was exposed in the electrolyte at its open circuit potential for 1 min,and then polarized at different current densities galvanostatically for 2 h.
Potentiodynamic polarization curves of the Mg were also measured in the three-electrode electrolytic cell containing 0.1 M HCl solution.The polarization potential scan started from -2.8 V to 1 Vvs.Ag/AgCl at 5 mV/s.
To evaluate the cathodic hydrogen reaction activity on the Mg surface after anodic polarization,the Mg electrode was firs galvanostatically polarized at an anodic current density for 10 mins,then potentiostatically polarized at a cathodic potential −2.2 Vvs.Ag/AgCl for 10 min,and anodically polarized in galvanostatic mode again at a more positive anodic current density for 10 min.The alternate galvanostatic anodic-potenostatic cathodic step polarization continued cyclically as the galvanostatic anodic current densities increased step by step from 5 mA/cm2to 60 mA/cm2.
2.3.Hydrogen evolution collection
Hydrogen was collected using a simple burette-funnelbeaker setup over the Mg electrode.A platinum counter electrode and an Ag/AgCl or Hg/Hg2SO4reference electrode were placed in a beaker containing 800 mL of the testing solution,and the hydrogen evolved from the polarized Mg working electrode under a funnel was led into a burette originally fille with the solution.
2.4.Analysis of surface morphology
To in situ observe the surface changes of the pure Mg electrode,an optical Leica Microscope (DVM-6a) was employed.The surface roughness of the corroded Mg electrode was analyzed using a Laser Confocal Scanning Microscope(VK-X200) and VK analysis software.The cross-sections of the corroded Mg samples were observed using Scanning Electron Microscopy(SEM,LEO-1530)with a secondary electron detector at working voltage 20 kV.The anodically polarized Mg surface was firstl sealed in epoxy resin,cut to expose its cross-section,then mechanically abraded using SiC paper up to 2000 grit,and finall sputtered with gold.
To further test the stability of the dark corrosion products or black layer in the corroding areas,an ethanol immersion test was carried out.The Mg specimen was firs galvanostatically polarized at 80 mA/cm2in 0.1 M HCl solution for 5 min to produce a large amount of the black substance or a large dark region on the Mg surface,then rinsed with high purity water quickly to remove the acid solution but still kept the black areas unchanged in color,and finall immersed in pure ethanol (99.8%) for 6 days to further observe the change of the black substance or the black area in color.
Each the experiment was repeated at least 3 times in this study.All the tests were carried out at room temperature,and all the potentials if not specifie in the paper were relative to the KCl-saturated Ag/AgCl reference.
3.Results
3.1.Hydrogen evolution
Figures 1a,1c,and 1e show the hydrogen evolution processes during the galvanostatic polarization tests in the 0.01 M,0.1 M and 0.2 M HCl solutions,respectively.The evolved hydrogen volume always increased with time.Figures 1b,1d,and 1f illustrate the corresponding potential variations.It seemed that most of the potentials did not change signifi cantly with time,except at a couple of large anodic current densities,indicating that the Mg surface during anodic dissolution might change evidently when anodically polarized at a high current density.
The same phenomena were also observed in a H2SO4solution.Fig.2a shows the hydrogen evolution and polarization potential variation under galvanostatic polarization at different anodic current densities in the 0.05 M H2SO4solution.The evolved hydrogen volume increased steadily with time,while most of the corresponding potentials were steadily unchanged with time,except that one continuously increasing at 40 mA/cm2.
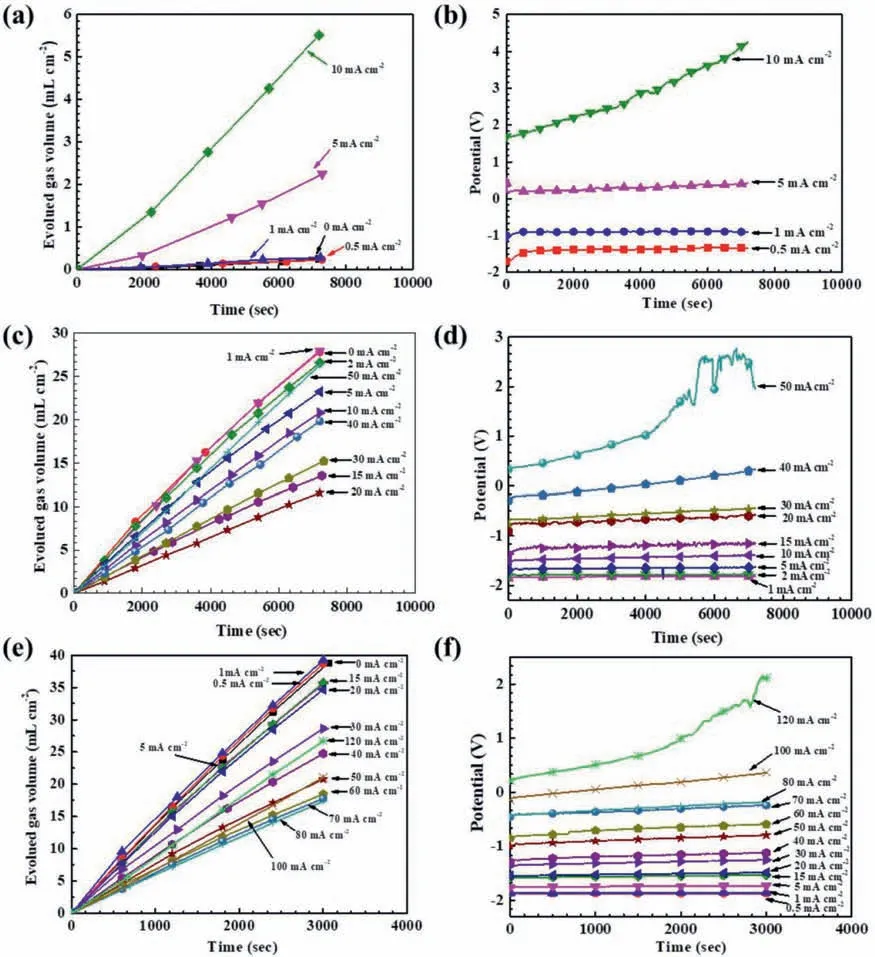
Fig.1.Galvanostatic polarization results for pure Mg at different anodic current densities in the HCl solutions with different concentrations: (a) hydrogen evolution in 0.01 M HCl,(b) polarization potentials in 0.01 M HCl,(c) hydrogen evolution in 0.1 M HCl,(d) polarization potentials in 0.1 M HCl,(e)hydrogen evolution in 0.2 M HCl,(f) polarization potentials in 0.2 M HCl.
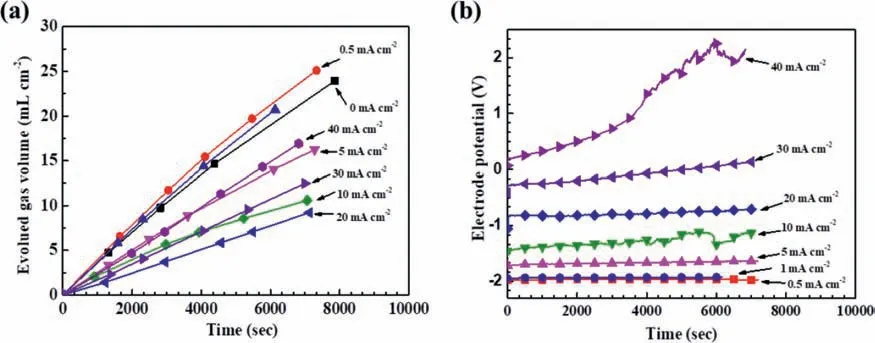
Fig.2.Galvanostatic polarization results for Mg at different anodic current densities in the 0.05 M H2SO4 solution: (a) hydrogen evolution curves,and (b)corresponding polarization potentials.
3.2.Surface morphology
Fig.3 shows the in situ surface morphologies of the Mg electrodes in the 0.1 M HCl solution under galvanostatic polarization at different anodic current densities.When the anodic polarization current densityI <20 mA/cm2,the Mg surface remained in silver color and the number of hydrogen bubbles from the Mg surface gradually decreased with increasing anodic current density (see Fig.3a,3b and 3c).IfI=20 mA/cm2,some dark regions appeared around the edges of the sample,possibly due to the relatively high current densities there.Meanwhile,the number of hydrogen bubbles decreased to the minimum (see Fig 3d).WhenI >20 mA/cm2,the total dark area increased with increasing current density until the whole surface became dark,and the density of hydrogen bubbles also increased with increasing anodic current density (see Fig 3e,3f and 3g).A close in situ observation indicated that the hydrogen mainly evolved from those dark regions,while on the silver regions the hydrogen evolution was relatively insignificant When the sample was polarized at a high anodic current,e.g.,80 mA/cm2for 20 min,the dark regions gradually turned into white,initially along the edges and then spreading to the whole surface (see Fig 3i).

Fig.3.The in situ surface morphologies of Mg electrodes immersed in the 0.1 M HCl solution at (a) open-circuit potential,under galvanostatic polarization at different anodic current densities (b) 0.5 mA/cm2,(c) 10 mA/cm2,(d) 20 mA/cm2,(e) 30 mA/cm2,(f) 40 mA/cm2,(g) 60 mA/cm2 and (h) 80 mA/cm2 for 2 min,and at anodic current density (i) 80 mA/cm2 for 20 min.
It was interesting,as shown in Fig.4,that the dark regions could be dissolved and regenerated,attaining a dynamic equilibrium in the edge areas for a long time.
Fig.5 presents the in situ surface morphologies of the Mg specimens immersed in the 0.05 M H2SO4solution under galvanostatic polarization at different anodic current densities,The surface changes with time were similar to those shown in the 0.1 M HCl solution (see Fig.4).AtI=20 mA/cm2,the dark regions were also firs formed along the surface edges and the hydrogen evolution rate decreased to the minimum (see Fig.5d).When the applied anodic current density continued to increase,the hydrogen evolution rate together with the dark regions increased.

Fig.4.The in situ surface morphologies of Mg electrodes immersed in the 0.1 M HCl solution under galvanostatic polarization at anodic current density 20 mA/cm2 for (a) 1 min,(b) 5 min,(c) 10 min,(d) 30 min,(e) 60 min and (f) 100 min.

Fig.5.The in situ surface morphologies of Mg electrodes immersed in the 0.05 M H2SO4 solution at(a)open-circuit potential,under galvanostatic polarization at different anodic current densities (b) 0.5 mA/cm2,(c) 10 mA/cm2,(d) 20 mA/cm2,(e) 30 mA/cm2,(f) 40 mA/cm2,(g) 60 mA/cm2 and (h) 80 mA/cm2 for 2 min,and at anodic current density (i) 80 mA/cm2 for 20 min.
It should be noted that some different hydrogen evolution behaviors were observed in the different surface regions at high anodic current densities (Figs.3 and 5).The hydrogen evolution in the dark regions was more intense than that in the silver regions.In general,the cathodic hydrogen evolution under potentiostatic cathodic polarization mainly came from the uncorroded area,and the anodic dissolution of Mg was closely associated with the anodic hydrogen evolution from the dark corroding areas under galvanostatic anodic polarization.
3.3.Anodic dissolution products on the surface
The dark corrosion products or the dark corroding regions on the Mg surface as shown in Figs.3-5 in summary had the following features:
(1) They could be dissolved in an acidic solution if the Mg electrode was not anodically polarized.
(2) They could sustain for a short period of time in a deareated or areated neutral aqeous solution,but would eventually turn into white.The dark region was unstable,and could not be easily measured by the current techniques,but the white fina corrosion products have been detected by XRD to be Mg(OH)2(see Appendix S1).
(3) They were relatively stable and could keep unchanged in pure ethanol,but some of the dark surface product layer could be fla ed off,as pointed out by the arrows in Fig.6.
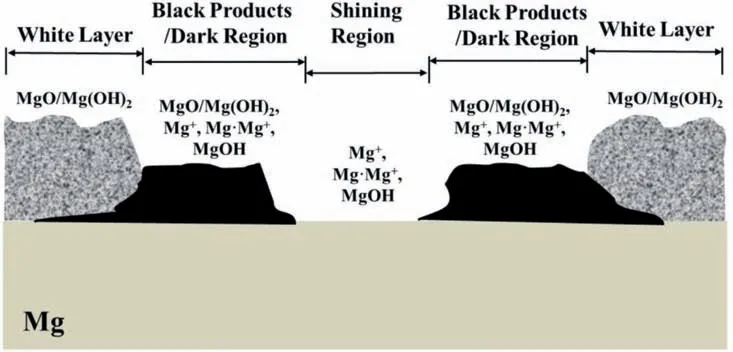
The surface morphologies of the Mg surfaces in air after mechanical abrasion and galvanostatic anodic polarization are shown in Fig.7.The mechanically abraded Mg surface was shiny and fla (Figs.7a,7a1 and 7a11),while the surface after galvanostatic anodic polarization at 30 mA/cm2consisted of shining regions in the central area and dark regions along the edges (Fig.7b),much rougher (Figs.7b1,7b11,7b2 and 7b21) than the mechanically abraded one.The dark regions along the surface edges appeared to be covered by a layer of rough,loose and black surface products.After galvanostatic anodic polarization at 60 mA/cm2,the initially shining central areas became dark and the initially dark edge areas turned into white (Fig.7c).The central areas also became rougher while turning into dark and also appeared to be covered by a layer of loose and rough surface products (Figs.7c1 and 7c11).The regions along the edges after turning from black to white were not shining,and looked like a thick layer of rough and loose surface products(Fig.7c21).It should be noted that hydrogen evolution was mainly from the dark regions either in the central or the edge regions.After they became white,the hydrogen evolution ceased there,while another area started to become dark and give out hydrogen.The surface roughness varied greatly from region to region (Figs.7c1,7c2 and 7c3).Generally,all the black products would become thicker and whiter gradually with time.The higher anodic current density applied,a thicker white surface layer would be eventually produced.

Fig.7.The surface morphologies of the Mg electrode in air.(a) The overall surface appearance of the Mg after abrasion with SiC paper up to 2000 grit finish (a1) The micro optical metallographic image of the area 1 in (a).(a11) The surface roughness of the area 1 in (a).(b) The overall surface appearance of the Mg after galvanostatic anodic polarization at 30 mA/cm2 in the 0.1 M HCl solution for 20 min.(b1) and (b2) The micro optical metallographic images of the areas 1 and 2 in (b),respectively.(b11) and (b21) The surface roughness in the areas 1 and 2 in (b),respectively.(c) The overall surface appearance of the Mg after galvanostatic anodic polarization at 60 mA/cm2 in 0.1 M HCl solutions for 20 min.(c1),(c2) and (c3) The micro optical metallographic images of the areas 1,2 and 3 in (c),respectively.(c11),(c21) and (c31) The surface roughness of the areas 1,2 and 3 in (c),respectively.
The cross-sections of the central areas of the pure Mg surfaces after galvanostatic anodic polarization in 0.1 M HCl solution for about 20 min are shown in Fig.8.After polarization at 60 mA/cm2,the surface corrosion product layer was uneven but loose,30–40 μm thick in average (Fig.8a),preferentially formed in some regions,leaving some areas uncorroded (Fig.8b).When the anodic galvanostatic polarization current density was 30 mA/cm2,the surface was very uniform,and no corrosion product layer was formed (Figs.8c and 8d).These results were in good consistence with those presented in Fig.3,further confirmin that in the dark regions there were anodic dissolution products deposited on the surface and the formation could be related to a high anodic current density.
3.4.Galvanostatic-potentiostatic behavior

Fig.6.The in situ surface morphologies of the Mg electrodes immersed in pure ethanol in different time after 5 min of galvanostatic anodic polarization at 80 mA/cm2.
To examine the catalytic activity for cathodic hydrogen evolution on an anodically polarized Mg surface,the Mg after galvanostatic polarization at different anodic current densities was immediately potentiostatically polarized at a cathodic potential −2.2 Vvs.Ag/AgCl.The selected galvanostatic anodic and potentiostatic cathodic conditions relative to the typical potentiodynamic polarization curve of the Mg specimen in the 0.1 M HCl solution are shown in Fig.9a.The changes of the galvanostatic anodic potentials after IR drop correction and the potentiostatic current densities with time are presented in Figs.9b and 9c,respectively.As the galvanostatic anodic current density increased step by step,the corresponding anodic potential in the step increased (Fig.9b),which had a similar trend to that presented in Fig.1d.The most important result was that the cathodic current density at the cathodic potentiostatic potential −2.2 Vvs.Ag/AgCl decreased as the galvanostatic anodic current density in the previous step increased (Fig.9c).Fig.6d shows the anodic and cathodic hydrogen evolution rates at as a function of the galvanostatic anodic current density.The measured anodic hydrogen evolution rate during the galvanostatic anodic polarization firs decreased and then increased as the applied galvanostic anodic current density increased.The cathodic hydrogen evolution rate measured during the step of the potentiostatic cathodic polarization immediately following the galvanostatic anodic polarization decreased as the previous galvanostatic anodic current density increased.
Fig.10 shows that some of the dark products formed on the Mg surface during the galvanostatic anodic polarization could be gradually dissolved in the immediate following step of the potentiostatic cathodic polarization,but some were unchanged,particularly after the galvanostatic polarization at a high anodic current density.This implies that the dark products could not result from cathodic hydrogen evolution.
4.Discussion
4.1.Hydrogen evolution under anodic polarization
The hydrogen evolution rates can be calculated from the average slopes of the hydrogen evolution curves shown in Fig.1,which are presented in Fig.11a.Similar to the observation made in the galvanostatic anodic-potentiostatic cathodic step polarization (see Fig.6d),there are also minimum hydrogen evolution rates at certain current densities in Fig.11a.In the 0.1 M HCl solution,the hydrogen evolution rate decreased with increasing anodic current density whenI <20 mA/cm2.AfterI >20 mA/cm2,it increased,exhibiting a typical NDE behavior or anodic hydrogen evolution characteristic.A minimum hydrogen evolution rate was reached at anodic current density 20 mA/cm2.As the acidic solution became more concentrated,e.g.,in the 0.2 M HCl solution,the hydrogen evolution rate became higher,but deceased more significantl with increasing anodic current density before reaching the minimums.The minimum hydrogen evolution rate shifted positively in a more concentrated acid(see Fig.11a).While the hydrogen evolution rates were increasing with increasing solution acidity at current densities negative to the minimum points,the hydrogen evolution rates at current densities more positive than the minimum points were not significantl influenced fl wing the same increasing trend with increasing current density (see the dash line in Fig.11a).
It should be noted that the minimum hydrogen evolution rate in experiment corresponded to the appearance of the firs dark corroding region on the Mg (Fig.3d).In fact,the formation of the dark products closely related to the onset of the accelerated anodic hydrogen evolution was more clearly visualized in situ in all the experiments in this study,but not very well illustrated in photos shown in Figs.3,5 and 7.The positive shift of the minimum hydrogen evolution rate in a more concentrated acid solution (Fig.11) had been observed in experiment to correspond to a black region later appearing on the Mg surface,which implied that the formation of the dark region or the black corrosion products was suppressed in a more acidic solution.
The hydrogen evolution rate as a function of the applied anodic current density in the 0.05 M H2SO4solution had a changing trend similar to that in the 0.1 M HCl solution (Fig.11).The minimum hydrogen evolution and the emergence of the firs dark regions on the sample surface also occurred at 20 mA/cm2.These two acids had the same normal concentration (0.1 M).However,at the same anodic current densities,all the hydrogen evolution rates in the 0.05 M H2SO4solution,were lower than those in the 0.1 M HCl solution.This confirm that Cl−is more corrosive than SO42−to Mg[23,40].
In this study using acidic solutions,no protective surface fil could be formed or the formed fil was too porous to be significantl resistant or protective.Thus,the surface fil or corrosion product layer had a limited contribution to the IR-drop in electrochemical experiments,and the solution resistance became the main influencin factor.In galvanostastic mode,the IR-drop could be easily estimated with applied current and known solution resistance.After IR-drop compensation,the hydrogen evolution rates in Fig.11a can be redrawn against IR-corrected polarization potentials in Fig.11b,in which the replotted curves also exhibit minimum hydrogen evolution rates;the minimum point positively shifts slightly with increasing acidity,and the hydrogen evolution rate monotonically increases continuously as the IR-corrected potential becomes more positive than 0.22 Vvs.Ag/AgCl (6a or 6b).It should be noted that this potential is very close to the equilibrium potential of the normal cathodic hydrogen reaction.If the hydrogen evolution under the anodic polarization is governed by a normal cathodic reaction (6a or 6b),the result will lead to a paradox that the cathodic hydrogen reaction can still be accelerated when the over-potential across the Mg/solution interface becomes more positive than the equilibrium potential of cathodic hydrogen reaction (6a or 6b) or the real driving force for the cathodic hydrogen reaction decreases to zero.This is unacceptable.

Fig.8.The SEM images of the cross-sections of the central areas of the pure Mg surfaces after galvanostatic anodic polarization at (a) 60 mA/cm2 and (b)30 mA/cm2 in the 0.1 M HCl solutions for 20 min,and the enlarged areas as indicated in (a) and (b) are shown in (c) and (d),respectively.
Even in the case that the IR-drop is incompletely compensated due to the coverage of a surface fil or some corrosion product deposits on the surface or a pit deep into the surface,the cathodic hydrogen evolution rate cannot increase with increasing polarization potential,particularly when the potential approaches the equilibrium hydrogen potential.This is because the real over-potential or the potential drop across the double electric layer at the Mg/solution interface as the driving force for the cathodic hydrogen reaction (6a or 6b)always becomes smaller as the applied potential increases.This can be demonstrated by the potential distributions over the Mg surface covered by a porous surface fil as illustrated in Fig.12.In the illustration,it is assumed that there is a pore in the surface fil in a selected surface region.Anodic Mg dissolution and cathodic hydrogen evolution occur on the exposed metallic Mg surface in the pore.Under anodic polarization,an anodic current fl ws from the Mg surface to the bulk solution in the pore through the film while cathodic current fl ws from the bulk solution to the Mg through the pore if the Mg is cathodically polarized.When the potential drop from the fil surface to the bulk solution is completely offset,the following potentials,resistances and current densities should be considered:
VM: the potential of metallic Mg,
VPL: the potential of the solution next to the metallic Mg surface in the fil pore,
VS: the potential of the bulk solution next to the fil surface,
E: the polarization potential or the potential drop between the metallic Mg and the solution bulk,i.e.,E=VM -VS,
EDL: the potential drop across the double electric layer at the Mg/solution interface in the fil pore,i.e.,EDL=VMVPL,
RSL: the solution resistance in the pore,
IPL: the current density following through the fil pore,and
IPLRSL: the IR-drop of the solution in the pore from Mg surface to the fil surface,i.e.,IPLRSL=VPL-VS.
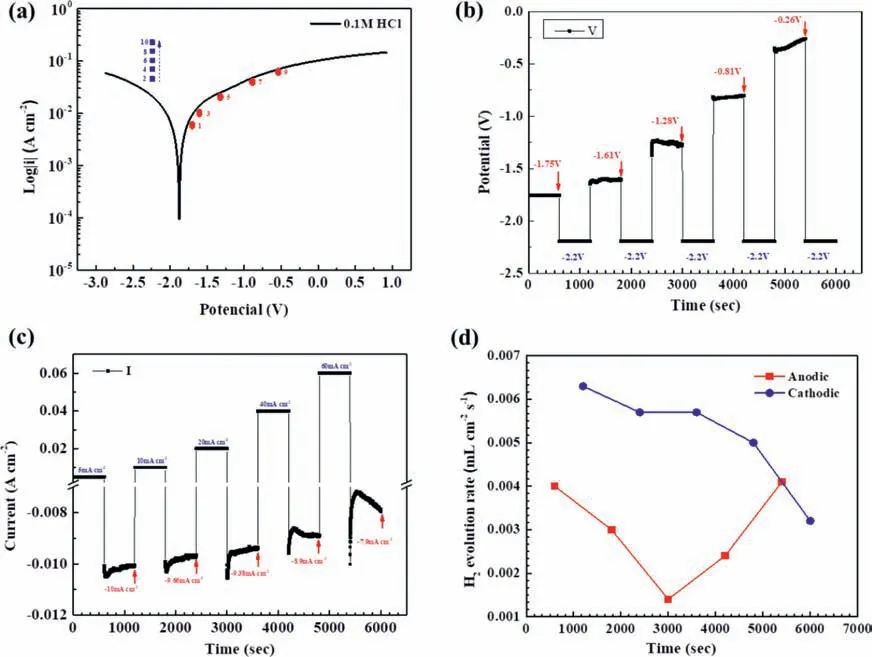
Fig.9.The potentials,current densities and hydrogen evolution rates of the Mg specimen under alternate galvanostatic anodic-potentostatic cathodic step polarization in the 0.1 M HCl solution: (a) the typical potentiodynamic polarization curve,the galvanostatic anodic potentials and corresponding current densities,and the applied potentostatic cathodic potentials and corresponding current densities in the immediate following steps,(b) the IR-corrected anodic potentials under the galvanostic polarization at the different anodic current densities as showed in (a) and the potentiostatic cathodic potential −2.2 V vs.Ag/AgCl in the immediate following steps,(c) the applied galvanostatic anodic current densities and the potentiostatic cathodic currents densities at cathodic potential −2.2 V vs. Ag/AgCl in the immediate following step,and (d) the hydrogen evolution rates under the galvanostatic polarization at different anodic current densities and under the potentostatic polarization at cathodic potential −2.2 V vs. Ag/AgCl in the immediate following steps.

Fig.10.The in situ surface morphologies of the Mg electrode in the 0.1 M HCl solution during the alternate galvanostatic anodic -potentiostatic cathodic step polarization (the photos were taken after 5 min in each galvanostatic anodic step).
It is well known that the anodic Mg dissolution and cathodic hydrogen evolution are determined byEDL;the anodic Mg dissolution increases while the cathodic hydrogen evolution decreases as theEDLincreases or becomes larger or more positive.The potential drop (VPL-VS) of the solution in the pore from the Mg surface to the fil surface is the driving force for theIPL.It is normal that all theEPL,ESLandIPLincrease with increasingE.A higher or more positiveVwill have a larger or more positiveEandEDL,which will result in a larger or more positiveIPLifRSLis a constant.When the Mg is polarized from a cathodic potential (e.g.,E1<0 in Fig.12) through the open circuit potential (i.e.,EOCPin Fig.12) to the equilibrium hydrogen potential (i.e.,EeHin Fig.12),which is an anodic potential(i.e.,E3 >0 in Fig.12),there will be:
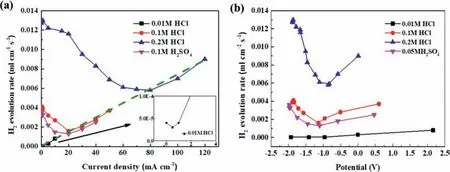
Fig.11.The hydrogen evolution rates at different (a) galvanostatic anodic current densities and (b) corresponding IR-corrected polarization potentials in 0.01 M,0.1 M,0.2 M HCl solutions and 0.05 M H2SO4 solutions.
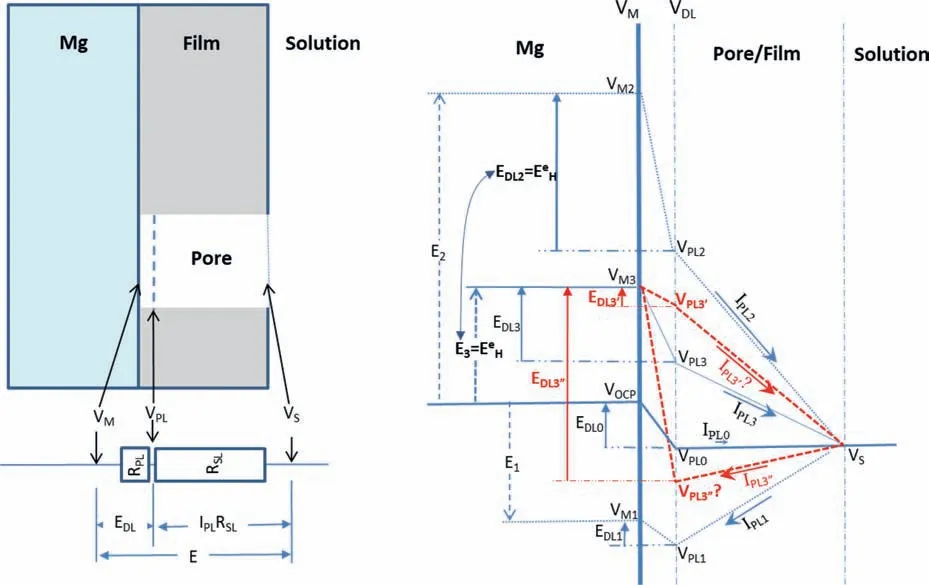
Fig.12.Schematic illustration of the potential distributions from the metallic Mg surface through a pore of the surface fil on the Mg to the bulk solution under different polarization conditions.
whereVOCPis the corrosion potential.Corresponding to the polarization potentialsE1,EOCPandE2,theV1,VOCPandV2are the potentials of the metallic Mg surface under the filmVPL1,VOCPandVPL2are the potentials of the solution in the pore next to the Mg surface,EDL1,EOCPandEDL2are the potential drops across the double electric layer at the Mg/solution interface in the pore,andIPL1,IPL0andIPL2are the current densities fl wing in the pore from the Mg surface to the bulk solution.It should be noted that at theV2or under theE2,
the hydrogen reaction is at its equilibrium state.Thus,there is no net cathodic hydrogen evolution.However,the anodic hydrogen evolution rate is remarkably high according to the mechanism of incomplete fil Mg+dissolution,because the anodic Mg dissolution is significantl fast.
In experiment if the IR-drop from the fil to the bulk solution can be compensated,but the IR-drop in the pore of the fil cannot,when a anodic polarization potentialE3,equal toEeH,lower thanE2(see Fig.12),i.e.,
is applied to the Mg,there will be:
Therefore,the anodic Mg dissolution and the resulting anodic hydrogen evolution will be strikingly faster than those at theEOCP,but lower than those at theE2.In this case,the cathodic hydrogen evolution should be much slower than that at theE2,but faster than that atEOCP.Correspondingly,the current density in the pore will be anodic:
All these have been experimentally observed and are reasonable according to the mechanism of incomplete fil Mg+dissolution.
However,according to the mechanism of enhanced catalytic activity,the cathodic hydrogen evolution rate atE3should be higher than that atEOCP,then theEDL3’E corresponding potential drop across the double electric layer at the Mg/solution interface must be more negative or lower than theEDL0(see Fig.12):
which will lead to the anodic Mg dissolution atE3slower than that atEDL0.The slower anodic Mg dissolution and the faster cathodic hydrogen evolution will produce a larger cathodic current density following from the bulk solution to the Mg through the pore:
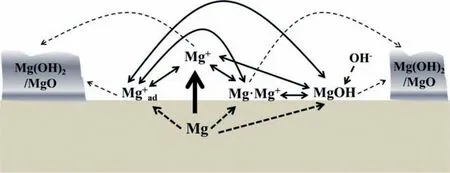
This contradicts 0 This can only result in a cathodic hydrogen evolution much slower than that atEOCP,which obviously cannot explain the enhanced cathodic hydrogen reaction by anodic polarization. Although the above analysis is based on a pore in the surface fil on Mg,it can be applied to a pit deep into the surface.The conclusion suggests that even when the IR-drop of the surface fil or a local pit cannot be compensated,the enhanced hydrogen evolution by anodic polarization,particularly when the polarization potential is approaching or above the equilibrium hydrogen potential,can only be explained by the mechanism of incomplete fil Mg+dissolution.It is impossible that the cathodic hydrogen evolution rate can increase as the polarization potential becomes more positive simply because of the uncompensated IR-drop of the surface fil on Mg or a local pit in Mg.In short,the increasing anodic dissolution current density with increasing potential means that the actual potential across the Mg/fil interface (or the overpotential determining the anodic current density) is becoming more positive.There is no way that an increased polarization potential can lead to an larger IR drop,but a decreased anodic current density,vice versa.Even if the IR-drop across the fil is increasing with the increasing anodic polarization potential or anodic current density,it cannot lead to faster cathodic hydrogen evolution,or slower anodic hydrogen evolution.The accelerated hydrogen evolution by anodic polarization can only be a result of reaction (4a,b,or c). The presence of a catalyst on the Mg surface may change the cathodic hydrogen reaction activity,but it cannot change the reaction thermodynamic trend.Therefore,Fig.11b strongly suggests the hydrogen evolution accelerated by anodic polarization cannot be a cathodic process.This further reinforces the conclusion based on similar hydrogen evolution results for Mg in a neutral NaCl solution in a previous study[15]. In fact,the hydrogen evolution rates of Mg in the acids firs decreased and then increased with increasing polarization potential or current density shown in Fig.11 were consistent with Rossruckeret al.’s wrok[41]on Mg anodically polarized in buffered solutions (pH=3).Fajardoet al.’s [42] measured hydrogen evolution on pure Mg under galvanodynamic anodic polarization in a 0.1 M citric acid solution buffered at pH=3 and also found that the hydrogen evolution rate increased with increasing current density.It was indicated that the enhanced hydrogen evolution rate and Mg dissolution by anodic polarization could be associated with the anodic dissolution of the exposed substrate Mg to Mg+at the surface fil breaks or in the pores of the corrosion product layer[14].They [12,27] in situ observed the growth of the corroding areas in dark color on the Mg surface accompanied by hydrogen evolution.The hydrogen evolution from the corroding areas was accelerated by anodic polarization and retarded or even stopped by cathodic polarization,while the hydrogen evolution in the cathodic regions or non-corroding areas ceased with increasing anodic polarization potential and was accelerated by cathodic polarization.With these observations and based on the model of incomplete fil Mg+dissolution[12,14],the notions of anodic hydrogen evolution (AHE) and cathodic hydrogen evolution (CHE) were proposed [14,27].It should be noted that the AHE can also take place at a cathodic potential,while the CHE can only occur at a potential more negative than the equilibrium hydrogen potential,which is not necessarily cathodic to the OCP. It appears that all the results in this paper can be interpreted by the AHE theory. In fact,the two different trends of the dependence of hydrogen evolution rate on polarization potential or current density (see Fig.11) suggest that two different mechanisms are governing the hydrogen evolution process: anodic hydrogen evolution (AHE) and cathodic hydrogen evolution (CHE)[12,27],as asserted in the model of incomplete fil Mg+dissolution [14].The AHE is a result of chemical reaction of water with the Mg+(4a,4b or 4c) generated by anodic dissolution(2)of Mg involving one electron.These reactions occur in the metallic Mg surface at the fil breaks or in the pores of the corrosion product layer,i.e.,the dark corroding areas observed in experiments.The CHE may also take place in the corroding areas and some cathodic phases in non-corroding areas. The experimentally measured total hydrogen evolution current density (IH) will be the sum of the AHE current density(IHa) and CHE current density (IHc),respectively,which can be written as: Correspondingly,according to the basic electrochemical principle,the net polarization current densityIwill be: According to the Butler-Volmer equation and the mechanism of incomplete fil Mg+dissolution,AHE current density will increase and CHE current density will decreased with increasing applied anodic current density.The detailed deduction is presented in Appendix S2.There should be a minimum in hydrogen evolution current densityIHminatEmin: In this case,there are alsoIc→0 andI≈Ia.Thus,the relation between hydrogen evolution current density and applied anodic current density can be obtained as shown in Appendix S2: Eq.(15) very well explains the hydrogen evolution rate linearly increases with increasing current density,independent of the HCl solution concentration when the applied anodic current density is more positive than the minimum point,as shown in Fig.11. From the slope of the dependence ofIHonI,we have: Thus,thekcan be estimated to be: reasonably within the range of 0≤k≤1 as expected by the model of incomplete fil Mg+dissolution.In the model,(1+k)electrons are apparently involved in the overall anodic dissolution of Mg in reaction (5).Only when all the electrons generated from reaction (2) are participated in reaction (3) on the Mg surface,i.e.,k=1,can the apparent valence (1+k)reach its highest value 2.0.On the contrary,if all the active intermediate univalent Mg+ions migrate quickly away from the Mg surface,without participating in reaction (3),thenk=0,and the lowest apparent valence will be 1.0.So far,Almost all the reported apparent valences for Mg dissolution are between 1.0 and 2.0,corresponding to akvalue between 0 and 1 [43],as expected by the model of incomplete fil Mg+dissolution is reasonable. The alternate galvanostatic anodic-potentiostatic cathodic step polarization presented in Fig.9 followed exactly the published experiment that was originally designed to support the idea of enhanced catalytic activity [44].In that work,pure Mg was anodically and cathodically polarized alternately in the 0.1 M NaCl solution and the hydrogen evolution during the cathodic polarization was measured.The results showed that the anodic polarization generated dark regions on the Mg surface,which enhanced the catalytic activity for hydrogen evolution and thus a higher cathodic hydrogen evolution rate in the immediate following step of cathodic potential [44].However,the increased rate of the cathodic hydrogen evolution from the cathodically polarized Mg immediately after anodic polarization could also be caused by the significantl roughened Mg surface during the anodic polarization.In current study,Fig.9 shows that the cathodic hydrogen evolution rate under the cathodic polarization immediately after anodic polarization was not enhanced;a more strongly anodically polarized Mg surface did not produce more hydrogen at a cathodic potential in the immediate following step of cathodic polarization.This phenomenon was repeatedly observed many times in the study.The photos in Figs.10g and i show that the increased amount of the black products on the Mg surface after the anodic polarization did not lead to more hydrogen bubbles generated in the immediately following step of cathodic polarization,either.It should be noted that the Mg surface in the acid was dissolved uniformly,and the corrosion products formed on the surface during the anodic polarization could be dissolved in the immediately followed cathodic polarization.Hence,the difference in surface roughness before and after the anodic polarization was relatively small,and the surface roughening effect was insignificant Obviously,these results (Figs.9 and 10) cannot be interpreted by the enhanced catalytic activity mechanism,either. The cathodic activity immediately after anodic polarization(Figs.9 and 10) can be interpreted based on the concentration(CMg+) of Mg+ions or the exposed areas (θ) of the substrate Mg at surface fil breaks or in the pores of the corrosion product layer according to the model of incomplete fil Mg+dissolution.TheCMg+andθare larger at an anodic potential,but they can decrease at a lower potential,and even become insignifican if the potential is more negative than a critical value [12,14].Therefore,in a neutral solution according to Eq.(11),the hydrogen evolutionIHawill be nearly zero and theIHcwill mainly be determined by the surface roughness.A larger anodic current will result in a rougher surface,which will have a faster cathodic hydrogen evolution reaction in the immediate following step of cathodic polarization,as reported in literature [44].In the acid solution in this study,θ→1 under both anodic and cathodic polarization,CMg+will be mainly dependent on the potential.Therefore,the cathodic hydrogen becomes relatively stable (see Figs.9 and 10) according to Eq.(11).At a high anodic current,a thick layer of corrosion products could still be generated (see Figs.9 and 10),which might to some extent inhibit the cathodic reaction.Thus,the cathodic current might even slightly decrease (see Fig.9c). The fundamentally different interpretations for the cathodic hydrogen activity between the models of enhanced catalytic activity and incomplete fil Mg+dissolution stem from the different understandings of the changes of Mg surface by anodic polarization in these two models.In the mode of enhanced catalytic activity,the catalyst produced by anodic polarization facilitates the cathodic hydrogen evolution in the immediate following step of cathodic polarization.The model of incomplete fil Mg+dissolution believes that no catalyst is produced on the anodically polarized Mg surface,but the exposed substrate Mg at the fil breaks or in the pores of the corrosion product layer,which can have a contribution to the cathodic hydrogen evolution. Yuwonoet al.[24] proposed that intermediates Mg∗H and Mg∗OH could be formed on Mg surface in order to interpret the hydrogen evolution enhanced by anodic polarization.These intermediates are in nature consistent with the univalent magnesium ion actually very well support the model of incomplete fil Mg+dissolution,because they are different forms of the intermediate Mg+and the active ion in them is Mg+.Since multi-electrons transferred in the elementary steps are unlike,the complicated electrochemical reactions they proposed [24] are not considered in this study.It is quite possible that the so-called catalyst in the model of enhanced catalyst activity is in effect an intermediate specie univalent Mg+ion or unstable Mg+containing compounds that can only survive for a relatively short period of time at the surface fil breaks or in the pores of the corrosion product fil in the dark corroding regions on the Mg surface.Since Mg+or its containing compound or specie is so closely associated with the exposed substrate Mg,its amount may be represented by the exposed areaθof the active substrate Mg.The exposed substrate Mg can act as an active reactant or site for hydrogen evolution,which may be misunderstood as a catalyst in the model of enhanced catalyst activity.This possibility will be further discussed,and the possible immediate active species on the Mg surface will be clarifie in the following section. The black corrosion products in the dark regions have been reported to be a thick insulating oxy-hydroxides containing some chlorides that are less protective than the initially formed oxides in air [37].The firs in situ observation[12,14,27] of such dark surface areas spreading out with time was made on a Mg alloy in a NaCl solution,in which the development of the dark area was found to be accompanied by hydrogen evolution and could be accelerated by anodic polarization and stopped by cathodic poalrization.In the dark regions,intensive hydrogen evolution and rapid Mg dissolution occurred [12,18,27,36,45].The closely related rapid Mg dissolution and hydrogen evolution can only be explained by the model of incomplete fil Mg+dissolution,not the enhanced catalyst activity.It can be reasonably deduced that the univalent Mg+ions or the Mg+containing species or compounds are generated and present in the dark corrosion products or the dark regions [46,47]. According the model of incomplete fil Mg+dissolution(Fig.13),Mg should be firs electrochemically oxidized to monovalent Mg+ions through reaction (2),some of which will be further electrochemically oxidized to Mg2+ions,while the remaining will chemically react with water,releasing hydrogen gas [12].A real process can be much more complicated than these.First,there may be an equilibrium reaction between the free Mg+ions in the solution adjacent to the Mg surface and the adsorbed Mg+adions on the surface: Moreover,the possibility cannot be excluded that some of the Mg+ions are temporarily stored in an intermediate Mg+containing compound or specie for a short period of time.For example,a possibility is that the Mg+is present in the form of Mg.Mg2+on the Mg surface through a dynamic equilibrium reaction [48]: If OH−ions are present,the Mg+may also interact with it to form an intermediate specie MgOH on Mg: All the intermediate species Mg+,Mg+ad,Mg.Mg2+and MgOH as listed above contain essentially a Mg+cation.They may co-exist on the substrate Mg surface in the micro pores of the surface product layer for a period of time if oxidant or water is not sufficientl present. Fig.13.Schematic illustration of the Mg dissolution reactions during corrosion. Regardless of the form the intermediate Mg+ion takes,all these Mg+ions containing intermediate species can react rapidly with water through: It is the Mg+(not the other elements) involved in the compounds that reacts with water to produce hydrogen.The Mg+comes from the anodic dissolution of Mg (2).Therefore,the hydrogen generation by the above reactions (21)through (23) in nature is the same as the anodic hydrogen evolution (4a,4b or 4c),and thus can also be accelerated by anodic polarization.It must also be borne in mind that none of these intermediate species or compounds is a cathodic reaction product.They are consumed in the hydrogen evolution reaction (4a,4b,4c,21,22 or 23) irreversibly and cannot be reproduced from any other intermediate products involved in a cathodic hydrogen reaction,returning their initial forms.Therefore,they are not catalysts for the cathodic hydrogen reaction. The above further development of the incomplete fil Mg+mechanism regarding the mixture of the intermediate active species with corrosion products on Mg surface can be employed to understand the changes in the black product or dark region on Mg surface during anodic polarization (Fig.14). According to the model of incomplete fil Mg+dissolution and the above developments,reactions (4a,4b or 4c)and (21) through (23) not only produce hydrogen,but also generate OH−ions or Mg(OH)2deposition.In an acidic solution under strong anodic polarization,a large amount of Mg+ions and Mg+containing species/compounds can be produced through reaction (2).They can be in forms of free Mg+,Mg+ad,Mg.Mg+or MgOH,and mixed with the deposited Mg(OH)2to form dark products on the Mg surface.Therefore,the hydrogen will mainly come from these dark regions (see Fig.3),the assertion can be easily understood that the dark region on the Mg surface results from a mixture of the Mg+ions or the Mg+ion containing intermediate species/compounds with the corrosion products MgO/Mg(OH)2,since intensive hydrogen evolution and severe corrosion damage can usually be directly visualized in the dark region (see Figs.3 and 5),which has also been widely reported in other studies [14,27].In the study,the initial dark area was found to be distributed in high current density regions (Fig.3),because more univalent Mg+ions were produced there.For the same reason,a high anodic current density generated a more intensive hydrogen evolution in a more rapidly growing dark region (Figs.3–5).The mixture theory for the dark products in a dark region on the Mg surface can also explain why only Mg,O and OH could be detected[20]. The observation (see Fig.7) that the dark products on Mg could quickly turn to white in air could be interpreted as the further oxidation of the intermediate species/compounds through chemical reactions (3),(4a,4b or 4c),(21),(22)or/and (23).In the acids under anodic polarization in this study,because of the continuous production of Mg+ions(2) to compensate for the consumption by the consumption reactions,the mixture of the Mg+or Mg+containing species/compounds with the corrosion products can be maintained.Therefore,the dark products were found on the Mg surface all the time (see Fig.4).As this product layer became thicker with time,the newly produced Mg+ions could not reach the outer layer.Hence,the thick surface layer appeared to gradually turn to white (see Figs.3 and 5).In pure ethanol,due to the absence of water and oxygen,the Mg+or Mg+containing species/compounds mixed in the surface layer could not be consumed by reactions (3) and (4a,4b or 4c),(21),(22) or/and (23).Hence,the dark Mg surface could preserve for a relatively long time (see Fig.6). Obviously,a sufficientl thickened dark product layer,although porous,on the Mg surface can to some degree reduce the current density,resulting in a redistribution of current density and the concentration of the univalent Mg+ions on the Mg surface.Hence,the dark regions were found to be moving around on the Mg surface in the study (see Figs.3–5). Fig.14.Schematic diagram for the mixture of intermediate species with corrosion products in a dark region on Mg. In the alternate anodic-cathodic polarization experiment,a large amount of the dark products were generated under the strong anodic polarization(see Fig.10).After the polarization was shifted to a cathodic potential,the reaction (2) would significantl slow down.Thus,the supply of the intermediate Mg+ions became very limited.In this case,the dark products on the surface could not retain a large coverage (see Fig.10). If the black products in the dark region is assumed to be something containing catalyst for the cathodic hydrogen evolution,then the Mg with a nicely polished shining surface would not immediately have a significan hydrogen evolution rate under a strong anodic polarization condition,as such an initial surface did not have sufficien catalyst to immediately facilitate the cathodic hydrogen reaction.If a suffi cient amount of catalyst could be produced immediately under the anodic polarization,then its amount should increase with time under that polarization condition,because the produced catalyst could not be consumed by the cathodic hydrogen reaction that it was catalyzing.However,these predictions were not supported by the experimental observations(see Fig.3),in which the hydrogen evolution volume always increased linearly with time with a constant evolution rate all the time from the beginning.The results agreed well with the presence of the intermediate active Mg+or Mg+containing species,whose concentration can be constant at a given anodic condition when its generation (2) and consumption (3,4a,4b,4c,21,22 or/and 23) reaches a dynamic equilibrium. In summary,the black substance or the dark region on the Mg surface in this study was likely a mixture of the intermediate Mg+or Mg+containing species/compounds with the fina corrosion products MgO/Mg(OH)2. 1) In an acidic solution where the protectiveness of surface fil on Mg has become insignificant the hydrogen evolution rate firs decreases and then increases with positively shifting polarization current density or potential.The normal cathodic hydrogen reaction dominates the hydrogen evolution process in the potential or current density range negative to the minimum hydrogen evolution rate point.The cathodic hydrogen evolution rate increases evidently with increasing acidity of the solution.At polarization potentials or current densities more positive than the minimum point,the anodic hydrogen evolution is continuously accelerated by increasing polarization current density or potential,and insignificantl influence by the solution acidity.All these experimental phenomena can be theoretically described by the mathematic equations developed based on the model of incomplete fil Mg+dissolution. 2) The intermediate active univalent Mg+ion generated from anodic dissolution of Mg may be present on Mg surface in different forms or contained in various species/compounds,such as the Mg+adadsorbed on Mg surface,the Mg.Mg2+in the Mg surface layer,and/or the MgOH in the surface product layer,in addition to the free univalent Mg+in the solution adjacent to the Mg surface. 3) The temporary black product or the dark region on Mg can be formed under a relatively strong anodic polarization condition.It is likely to be a mixture of MgO/Mg(OH)2with Mg+ions and/or the Mg+containing species listed in conclusion 2).They will eventually become white after these intermediate active species turn into more stable Mg2+containing compounds,such as MgO/Mg(OH)2. 4) The IR-drop across the surface fil or from the metallic Mg to the reference electrode cannot change the changing tendency of the potential drop at the interface of Mg/solution with applied polarization potential.An increased IR-drop caused by a more positive anodic polarization potential or current density cannot lead to a slower anodic hydrogen evolution.As long as the anodically applied polarization current density or potential becomes more positive,the over-potential across the Mg/solution increases.Thus,the anodic hydrogen evolution rate always increases with increasing anodic current density or applied polarization potential.It is impossible for cathodic hydrogen evolution to be accelerated when the applied polarization potential or current density becomes more positive. Acknowledgments The research was supported by the National Science Foundation of China (key project grant No.51731008 and general project grant No.51671163). Supplementary materials Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.jma.2021.05.008.4.2.Interpretations based on cathodic and anodic hydrogen evolution

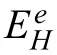
4.3.Surface catalysis or activation for hydrogen evolution
4.4.Surface intermediate active species for hydrogen evolution
5.Conclusions
 Journal of Magnesium and Alloys2023年1期
Journal of Magnesium and Alloys2023年1期
- Journal of Magnesium and Alloys的其它文章
- Development of high-strength magnesium alloys with excellent ignition-proof performance based on the oxidation and ignition mechanisms: A review
- Development and application of magnesium alloy parts for automotive OEMs: A review
- Recent advances in surface endothelialization of the magnesium alloy stent materials
- Recent developments in high-pressure die-cast magnesium alloys for automotive and future applications
- Exploring the contribution of oxygen reduction reaction to Mg corrosion by modeling assisted local analysis
- Microstructures,mechanical properties,corrosion,and biocompatibility of extruded Mg-Zr-Sr-Ho alloys for biodegradable implant applications