
CO2和甲醇直接合成碳酸二甲酯催化剂研究进展
2023-03-18 10:57:42李鹏阳张彩东么洪勇田志强王雪琦
工业催化 2023年2期
李鹏阳,张彩东,么洪勇,田志强,韩 星,王雪琦
(河钢材料技术研究院, 河北 石家庄 050023)
随着工业的快速发展,大量的CO2被钢铁、石化等企业不断排放到大气中,危害人类的生存环境。在“双碳”目标下,CO2作为C1原料转化为高附加值精细化学品是目前的研究热点,资源化利用CO2不仅能降低碳排放,还可带来经济效益,CO2与甲醇直接合成碳酸二甲酯(DMC)是有效途径之一[1]。
DMC是重要有机合成中间体,可用于生产聚碳酸酯或聚氨酯,作为电解液用于锂电池行业,因其较高的辛烷值可作为燃料添加剂,此外,DMC还被用作表面活性剂和抗氧剂等[2]。DMC的合成途径很多,包括光气法、甲醇氧化羰基化法、酯交换法和直接合成法等[3],其中CO2和甲醇直接合成DMC是研究最为广泛的合成路线,此反应直接消耗CO2且副产物只有水。目前,反应研究仍处于实验阶段,并面临许多挑战。该反应受热力学限制,催化剂的选择和设计最为关键,催化剂高效回收再生以及失活原因也是研究的重点。若能解决上述等问题,并实现直接合成DMC的工业化生产,将对碳减排具有重要的意义。本文总结了均相催化剂、非均相催化剂、脱水剂在CO2和甲醇直接合成DMC反应中的应用,并对不同催化体系下的催化作用机理进行介绍。
1 均相催化剂
1.1 离子液体催化剂
离子液体具有液体范围广、无挥发性、物理化学性质可调性好等特点,被广泛用于均相催化领域[4],但其回收困难且催化体系较复杂。

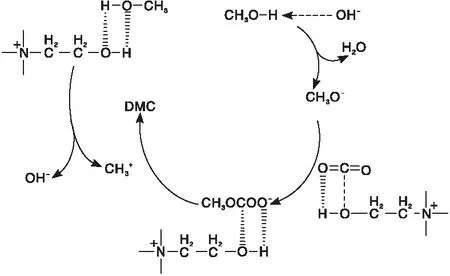
图1 离子液体的催化反应机理[5]Figure 1 Catalytic reaction mechanism of ionicliquids[5]
与羟基功能化离子液体相比,新型三元催化体系的催化性能更优,但催化体系较复杂。在1-(2-羟乙基)-3甲基咪唑双(三氟甲磺酰)亚胺([EmimOH][NTf2])、氧化氯化钐(DBU)和1,8-二氮杂环[2.2.2]十一-7-烯(SmOCl)组成的三元催化体系下,甲醇转化率为13.0%,DMC选择性高达99.13%[6]。研究发现,此体系也是通过OH-吸收CO2,甲醇则是被SmOCl所提供的Lewis酸性氯化物和Lewis碱性氧化物的协同组合进行激活。
Tamboli A H等[7]合成了质量分数10%壳聚糖/离子液体(IL)催化剂,溶解于离子液体的聚糖生物聚合物提供富电子的氨基和羟基源用于捕获CO2,IL使壳聚糖上的活性位点比固体壳聚糖上的活性位点更易接近,与三元催化体系相比,此体系更为简化且甲醇转化率从13.0%提高到16.9%,DMC收率达到16.68%。同时,该方法也为如何将丰富的生物聚合物溶解在离子液体中并直接利用富电子氨基或羟基源捕获CO2提供了一个示例。
[CnCmIm][HCO3]是一种具有脱水能力的咪唑型碳酸氢盐离子液体,其催化脱水机理如图2所示,[CnCmIm][HCO3]能吸附CO2并迅速转化为CnCmIm-CO2,且在催化体系中两者之间存在平衡,CnCmIm-CO2一方面促进DMC的生成,同时又与H2O反应降低热力学限制,在重复使用后催化性能和脱水性能无明显损失[8]。Pawar A A等[9]采用乙二醇等廉价原料合成了具有不同阴离子和相同阳离子的离子液体,其中EG[Vim]2[NTf2]2/二甲氧基丙烷(DMAP)催化体系的效率最高,在6.5 Mpa、130 ℃下反应6 h,DMC收率达41.9%,选择性为91.8%。反应结果表明,离子液体因其温和的酸碱性质具有较高的CO2吸收能力,在最佳反应条件下能更有效地实现甲醇的传质。

图2 [CnCmIm][HCO3]的催化脱水机理[8]Figure 2 Catalytic dehydration mechanism of [CnCmIm][HCO3][8]
1.2 碱性碳酸盐催化剂
碱性碳酸盐对催化CO2和甲醇直接合成DMC也具有活性,但对DMC的选择性偏低。在Li、Na、K、Cs、Mg等碱性碳酸盐中,K2CO3的催化性能最佳,催化过程需要CH3I的辅助,与HI反应生成KI会导致催化剂失活[10]。
有研究[11]表明,K2CO3并不起催化作用而是与反应物构成了一个耦合反应系统,打破反应的热力学限制。蔡振钦等[12]发现添加离子液体可改善K2CO3的催化性能,反应中添加溴代1-乙基3-甲基咪唑盐离子液体(EmimBr)后,DMC的生成速率加快,在90 ℃、3.6 MPa、甲醇/CH3I/K2CO3质量配比为12∶2.3∶1条件下,DMC收率可达6.50%,但离子液体的添加量并非越多越好,而是存在最佳值。
2 非均相催化剂
2.1 金属氧化物催化剂
金属氧化物不存在氧化失活,有很大的工业应用优势,主要以CeO2、ZrO2等过渡金属氧化物为主。较高的催化活性和稳定性使其成为目前研究最为广泛的催化剂,且复合金属氧化物相对于单一金属氧化物性能更优。
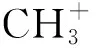

图3 ZrO2或CeO2上CO2和甲醇直接合成DMC反应机理[13-14]Figure 3 Reaction mechanism of DMC synthesis from CO2 and methanol over ZrO2 or CeO2[13-14]
催化剂形貌对催化性能有较大的影响,而且表面的酸、碱性位点均可促进DMC的生成,两者显示出协同效应[15]。Wu Zili等[16]认为纺锤状CeO2比棒状、立方体、八面体CeO2表现出更高的活性的原因与催化剂上的缺陷密度、酸碱位点数量以及CeO2(111)晶面有关。Unnikrishnan P等[17]的研究证明,纺锤状CeO2催化剂上的大量缺陷可吸附更多的CO2,并被中等强度的酸、碱性位点激活,因此更有利于生成DMC。Zhao Shuyang等[18]对比纳米棒、八面体、立方体三种形貌CeO2的催化性能,发现活性最好的纳米棒CeO2上具有更多的羟基官能团和双齿碳酸盐,而且CeO2的(110)晶面和(111)晶面对双齿碳酸盐的形成更有利,进而可生成甲基碳酸盐中间体。
采用磷酸(H3PO4)对金属氧化物进行改性可有效改变催化剂表面的酸性位,改性后催化剂中的Bronsted酸性位点可促进甲醇的活化[21]。Wu X L等[22]采用H3PO4改性V2O5,结果表明,催化剂上的Bronsted酸位点比Lewis酸位点对甲醇活化产生甲基更有效。磷酸对CeO2-ZrO2表面的处理也有报道,Prymak I等[23]发现经H3PO4改性后CeO2-ZrO2相组成发生了较大变化,形成了纳米尺度上的富Ce四方相和富Zr四方相的CexZr1-xO2,导致催化剂上Lewis酸性位点增加(不饱和表面Zr4+阳离子),使甲醇在反应中形成甲氧基中间体(Zr4+-OCH3)的能力增强,在170 ℃、6.5 MPa下反应1 h,DMC收率从0.24%提高至1.6%。
相对于单一金属氧化物,复合金属氧化物表现出更高的催化活性和化学稳定性,物种的掺杂可调变催化剂表面的酸碱性位点和氧空位。陈红萍等[24]发现α-Fe2O3-ZrO2催化剂中的α-Fe2O3和ZrO2主要为六方相和四方相晶型,双金属间的相互作用使催化剂表面的Lewis酸增强,并产生少量的Bronsted酸,Bronsted酸在活化甲醇产生甲基的同时又降低了速控步骤的能垒,更有利于DMC的生成。Li Aixue等[25]发现具有六方相和四方相Fe2O3共存结构的Fe0.7Zr0.3Oy混合金属氧化物有利于形成中等酸、碱性位点,对DMC选择性高达100%,研究发现,Fe的加入影响ZrO2的晶体结构,从而增加催化剂上的酸碱性位点,随着Fe含量的增加,比表面积显著增加,导致更多的活性位点暴露。CeO2纳米棒中掺杂TiO2也可增加催化剂表面的酸碱性位点,进而提高催化剂活性,初始速率法测定催化剂的表观活化能为46.3 kJ·mol-1[26],与报道的CeO2纳米棒(65 kJ·mol-1)和商业CeO2(117 kJ·mol-1)相比[27],催化剂表观活化能更低,故DMC的生成更容易,在140 ℃、1 MPa和360 h-1空速下,Ti0.04Ce0.96O2纳米棒于固定床反应器中催化CO2和甲醇反应,7 h后甲醇转化率达5.38%,DMC收率为4.47%。
CeO2基体中加入不同量的CaO可引起催化剂结构和表面性能的变化,而且CaO与CeO2的相互作用提高了表面氧空位的数量,增强了对CO2的吸附[28-29]。Liu Bin等[30]发现Zr掺杂的CeO2纳米棒具有更多的氧空位,原因在于Zr的掺杂使CeO2晶格中产生类似萤石的固溶体,从而促进氧空位的形成。CeO2上负载Cu也会增加氧空位的形成并提高其碱性,Cu2+取代了CeO2晶格中的Ce4+形成了具有碱性的氧空位,但并不是Cu的负载量越大越好,增加Cu的负载量会导致CeO2上CuO相沉积,而CuO相的存在导致副反应的发生[31]。Chen Yongdong等[32-33]研究了不同Ti和Zn掺杂量的MxCe1-xO2(M=Ti、Zn)纳米粒子包覆在蜂窝陶瓷上的整体式催化剂,当x=0.1时催化剂活性最高,整体式Ti0.1Ce0.9O2催化剂的活性几乎是相应颗粒催化剂的两倍,在140 ℃、2.4 MPa条件下,甲醇转化率为24.3%,DMC收率可达19.08%,但DMC选择性稍低。CeO2中掺杂Ti既可以增加活性位点,又能提高氧空位的浓度;Zn离子的掺杂使CeO2晶格中形成萤石结构的固溶体,改变了催化剂的结构和表面性质,使其具有更大的比表面积、更高的氧空位浓度,催化剂具有的规则直通道结构可及时去除水,在160 ℃、2.4 MPa条件下,甲醇转化率较前者有所降低,但DMC选择性增大,DMC收率达16.83%。
DFT计算可为设计CO2和甲醇直接合成DMC的有效催化剂提供分子水平的指导。Jiang Jian等[34]对CO2和甲醇在还原态CeO2(111)和CeO2(110)晶面上的反应进行了DFT计算,确定了以单甲基碳酸酯(MMC)为关键中间体,包括MMC的生成、酯化和水分子去除的三步朗格缪尔-欣谢尔伍德(L-H)机理,并根据能量分布比较,证明氧空位可显著降低MMC生成和酯化的活化能垒,在还原态CeO2(110)晶面,MMC的酯化具有最高的能垒,在还原态CeO2(111)晶面,水分子的去除是阻碍氧空位再生的限速步骤。通过计算结果指出催化反应中的两点不足:1)氧空位倾向于阻碍水分子的去除;2)甲醇分子倾向于占据氧空位,导致甲醇分子和CO2分子在被还原的CeO2表面上竞争催化位。
2.2 负载型催化剂
负载型催化剂在CO2和甲醇直接合成DMC反应中的应用也较为广泛,活性组分主要以双金属Cu-Ni为主,现存问题主要有商业载体成本高、活性低和寿命短等,所以合成新型载体是提高催化性能的有效途径。
钟顺和等[35]发现Cu-Ni/V2O5-SiO2(VSiO)催化剂表面有Cu-Ni合金活性中心、Lewis酸活性中心(Vn+)和Lewis碱活性中心(V=O),并提出如图4所示的反应机理,CO2在Cu-Ni合金和Lewis酸位的协同作用下生成吸附态二氧化碳卧式物种并解离为M-CO和V=O物种,甲醇在Lewis酸碱位的协同作用下形成V-OCH3和V-OH物种,M-CO和V-OCH3物种结合生成DMC。
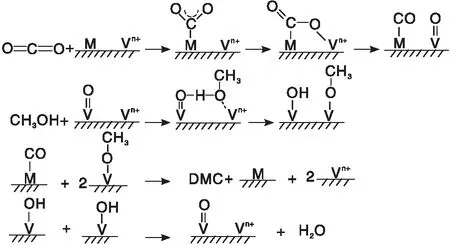
图4 CO2和甲醇在Cu-Ni/V2O5-SiO2上的反应机理[35]Figure 4 Reaction mechanism of DMC synthesis from CO2 and methanol over Cu-Ni/V2O5-SiO2[35]
载体的特殊结构以及金属颗粒与载体之间的强相互作用可提高催化剂活性[36]。Bian Jun等[37]以Cu-Ni合金为活性金属,设计并合成了钒(V)掺杂活性炭载体,钒的加入可增强活性金属颗粒在载体表面的分散性,还可抑制活性金属向低价金属的还原,提高催化剂的稳定性,在110 ℃、1.2 MPa条件下,DMC收率达7.01%。Chen Huiling等[38]将Cu-Ni合金负载于0.4 nm分子筛上,在固定床中催化CO2和甲醇直接合成DMC,在120 ℃、1.1 MPa、510 h-1空速下,5 h后DMC收率达5.0%,研究发现金属颗粒与载体之间存在相互作用,并对载体的酸碱性有明显影响,碱性位点促进甲醇活化为甲氧基物种,并在酸性位点上与活化的CO2反应生成DMC,但催化剂表面积随金属含量的增加而减小。Pimprom S等[39]通过溶胶-凝胶法,采用物质的量比50∶50的原硅酸四乙酯和稻壳灰制备的硅酸钠溶液合成了介孔二氧化硅(SBA-15),并负载Cu-Ni双金属。研究发现,载体为普通六边形结构,添加Mo进行改性后,催化剂的酸度、活性及稳定性得到提高, DMC收率由4.30%提高至5.04%。
郭林秀等[40]采用回流法制备了α-磷酸锆(α-ZrP)载体并负载2Cu-Ni双金属,结果表明,α-ZrP是具有层状结构、比表面积大、热稳定性高的理想载体,催化剂表面的酸碱性位有利于CO2和甲醇的活化,500 ℃下焙烧的(2Cu-Ni)/α-ZrP催化剂活性最好,在130 ℃、0.6 MPa条件下,DMC收率为4.68%。以多孔晶体沸石咪唑酯骨架-8(ZIF-8)为载体的Cu-Ni负载型催化剂同样具有良好的催化性能[41],提高反应温度可以提高甲醇转化率和DMC收率,但高于110 ℃的反应温度会破坏ZIF-8的骨架结构,而且对DMC的选择性偏低仅有50%,催化剂在回收和重复使用多次后性能略有下降。
相比于传统溶液法,采用新型硫化法合成的Cu-Ni/VSiO催化剂含有更多的合金相,且与CO2和甲醇的相互作用更强,活性可提高约3倍,原因在于新型硫化法极大改变了催化剂的微观结构,进而提高催化剂的性能,在120 ℃、1.2 MPa下,DMC收率为3.91%[42]。Chen Yongdong等[43]制备了具有热稳定的三苯基膦基多孔有机聚合物(POP-PPh3)载体,载体的多孔结构以及Cu-Ni合金纳米粒子在其上的均匀分布使催化剂具有较优的催化性能,在160 ℃、2.4 MPa下,DMC收率达8.40%。Arbelaez-perez O F等[44]考察了负载于活性炭的Cu-Ni双金属催化剂活性,当n(Cu)∶n(Ni)=2∶1时活性最佳,动力学研究表明,CO2与甲氧基物种的吸附为反应速率的决定步骤。
2.3 杂多酸催化剂
杂多酸具有特殊的空间构型、氧化还原性质、Bronsted酸位、“准液相”行为等特点[45]。目前,CO2和甲醇直接合成DMC的杂多酸催化剂主要有磷钨杂多酸、磷钼杂多酸以及经H3PW12O40修饰的金属氧化物,杂多酸催化剂的反应机理主要基于Bronsted酸性位理论和氧空位理论。
相比于无机酸,杂多酸的Bronsted酸强度更高,因此在催化CO2和甲醇直接合成DMC反应中具有更高的活性。Aouissi A等[46-47]对比了Co1.5PW12O40、Co1.5PMo12O40、Fe1.5PMo12O40、Fe1.5PW12O40四种keggin型杂多酸催化剂,其中Co1.5PW12O40的催化活性最好,其高活性得益于Co和W双金属间的协同作用。随着反应温度的升高,CO2在催化剂表面的吸附量减少,甲醇转化率和DMC选择性下降,原因在于DMC在Co1.5PW12O40上高温下分解,当反应温度大于200 ℃时,生成二甲氧基甲烷和甲酸甲酯。
Chiang C L等[48]通过溶胶-凝胶法制备了H3PW12O40/Ce0.1Ti0.9O2催化剂,并基于氧空位理论对催化反应机理进行研究,如图5所示,催化剂晶体结构缺陷,表面存在大量的氧空位,CO2分子的氧原子填补一个空位,两侧相邻空位则被甲醇分子的氢原子填补,结合产生不稳定的中间体,最后中间体分解为DMC和H2O分子并从氧空位中解吸,完成催化循环。该反应机理也从氧空位和晶体缺陷的角度解释了杂多酸催化活性的本质。利用H3PW12O40改性ZrO2催化剂可为其提供Bronsted酸性位点,且Bronsted酸性位点对甲醇活化的作用比Lewis酸性位点更强[22],在170 ℃、CO2/N2体积流量比为1∶7下,DMC收率为4.00%,根据阿伦尼乌斯方程估算得出催化剂的表观活化能为15.54 kJ·mol-1[49]。

图5 H3PW12O40/Ce0.1Ti0.9O2催化CO2和甲醇直接合成DMC的机理[48]Figure 5 Mechanism of direct synthesis of DMC from CO2 and methanol catalyzed by H3PW12O40/Ce0.1Ti0.9O2[48]
3 添加脱水剂的催化体系
CO2和甲醇直接合成DMC反应受热力学平衡限制,且生成的水可导致催化剂中毒失活。脱水可改变化学平衡,在提高甲醇转化率和DMC收率的同时,还可延长催化剂使用寿命。
研究发现腈类化合物是有效的脱水剂,其中2-氰基吡啶(2-CP)与CeO2组成的催化体系在固定床反应器中催化CO2和甲醇直接合成DMC,在120 ℃、20 MPa下,DMC收率达94.1%[50]。有研究者还发现2-CP中氰基碳的高电荷数有助于促进DMC的合成[51]。
2-CP水合产生的2-吡啶酰胺(2-PA)在CeO2上吸附后会毒害其上活性位点[52]。Stoian D等[53]在实验中通过肉眼观察到CeO2表面被一些结晶2-PA包裹,在略高于2-PA沸点的温度下进行原位甲醇洗涤和热处理,可将2-PA除去,实现催化剂的完全再活化。利用稀土金属(La、Gd、Pr)对CeO2表面改性可抑制2-PA的吸附,同时还能提高甲氧基物种的吸附,Ce4+物种的表面碱性和还原性在防止催化剂失活方面发挥着重要作用,其具有稳定活性甲氧基物种的能力[54]。
在均相或非均相催化体系中,4-二甲基氨基吡啶(DMAP)对CO2和甲醇直接合成DMC反应均显示出良好的脱水效果[55]。Pawar A A等[9]发现DMAP与乙二醇-二(1-乙烯基咪唑)二甲酰离子液体组成的催化体系对DMC的合成表现出良好的催化性能,和2-CP与H2O反应生成2-PA不同的是,DMAP通过碱性温和的氨基捕获水分子,除此之外,其还具有提供Lewis碱的作用,DMAP和离子液体的结合提供了DMC合成所需的中等Lewis酸性和碱性位点,可捕获更多的CO2,使平衡向产物生成方向移动,在130 ℃、6.5 MPa下反应6 h,DMC收率可达41.9%,同时也能保持着较高的选择性(91.8%)。
二甲氧基丙烷(DMP)是一种极易水解的缩酮,毒性较低。Wang Shengping等[56]对比了纺锤状、八面体、立方体三种形貌CeO2在有无脱水剂存在下的催化性能。结果表明,纺锤状CeO2活性最高,由于热力学限制,未添加DMP的情况下,三种催化剂的DMC收率都很低,随着DMP脱水剂的添加,DMC收率先增加后减小,原因在于添加大量DMP后甲醇浓度降低,DMP的最佳用量为3.6 mL,在140 ℃、2 MPa下反应24 h,DMC收率达59.3%,此时DMC在反应平衡体系中饱和,热力学计算表明,DMP的加入使反应平衡常数从6.3×10-3增加到25×10-3。Faria D J等[57]提出了一种新的温和条件下的脱水系统,即分子筛捕获气相中水与DMP捕获反应液相中水相结合的方式,在80 ℃、4 MPa下,甲醇转化率达48.6%,DMC收率达42.8%。
碳化钙(CaC2)也具有脱水能力,Zhang Zhaofu等[58]以二丁基氧化锡(Bu2SnO)为催化剂,首次将CaC2与CO2和甲醇直接合成DMC反应相结合。结果表明,CaC2与生成的H2O反应生成乙炔气体和Ca(OH)2,致使平衡向正方向移动,但CaC2不能重复使用,因为其已经转化为乙炔。CaC2还会与甲醇反应生成醇钙Ca(OCH3)2,醇钙经水解后生成Ca(OH)2和甲醇,在180 ℃、15 MPa下反应10 h,DMC收率达11.3%,而无CaC2加入时DMC收率仅有0.9%。
三氯乙酸甲酯(MTCL)作为脱水剂比2-CP具有更高的水解活性,以2%Cu/CeO2为催化剂,在140 ℃、5 MPa下反应3 h,与2-CP相比,MTCL作为脱水剂时的DMC收率高出其两倍以上,可达11.2%,但脱水过程中会副产甲缩醛和甲酸甲酯,造成DMC选择性降低,仅为80%[31]。
4 结语与展望
CO2和甲醇直接合成DMC是降低碳排放和实现碳资源利用的有效途径之一,对于将CO2转化为高附加值化学品以及实现绿色低碳可持续发展具有重要的现实意义。
然而,热力学平衡的限制、较低转化率和收率的技术挑战未得到根本解决,阻碍了工业化的发展。高效稳定的催化剂仍是目前研究的关键,迄今为止催化剂类型主要有离子液体、碱性碳酸盐、金属氧化物、负载型催化剂、杂多酸等,其中金属氧化物性能稳定,是目前研究最为广泛的催化剂,具有工业应用前景,其催化的本质核心是酸碱位和氧空位。脱水剂在CO2和甲醇直接合成DMC反应中的应用研究也取得了不错的进展。
笔者认为复合金属氧化物可作为工业化催化剂的首选,通过改性、掺杂物种等方式调变表面氧空位密度和酸碱位点数量,进而提高催化活性;开发能最优分散活性组分且具有吸收水效果的高效载体,并联合脱水剂使用,能及时、连续地消耗产生的水分子,降低热力学限制;深入催化反应机理的研究,结合DFT计算,确定反应的速控步骤,采取手段降低速控步骤的能垒。这些研究在今后的工作中值得进一步关注。
猜你喜欢
化工管理(2022年14期)2022-12-02 11:46:54
能源化工(2021年2期)2021-12-30 18:31:06
石油石化绿色低碳(2019年6期)2019-02-13 09:39:01
陶瓷学报(2019年5期)2019-01-12 09:17:38
广东石油化工学院学报(2016年6期)2016-05-17 05:17:25
中国化肥信息(2016年27期)2016-05-17 04:25:18
合成化学(2015年5期)2015-03-26 06:02:22
读者欣赏(2014年6期)2014-07-03 03:00:48
石油化工应用(2014年2期)2014-03-11 17:38:59
语文知识(2014年2期)2014-02-28 21:59:21
