
高错误率长序列基因组数据敏感序列识别并行算法
2023-03-16 00:58:38钟诚孙辉
通信学报 2023年2期
钟诚,孙辉
(1.广西大学计算机与电子信息学院,广西 南宁 530004;2.广西高校并行分布与智能计算重点实验室,广西 南宁 530004)
0 引言
基因组数据是一类重要的医疗大数据,被广泛应用于生物医学、定制化医疗服务、法医学鉴定和全基因组关联研究等领域[1-2]。基因组数据敏感序列是指可用于发起推断攻击、成员身份攻击、身份追踪攻击和完全攻击等基因组数据隐私攻击的核苷酸序列,包括短串联重复(STR,short tandem repeat)序列和疾病相关序列(DRS,disease-related sequence)[3]。不同于网络隐私数据的可重塑性,基因组数据敏感序列信息的泄露会对隐私泄露者的医疗保险、社会就业和公共福利等产生不利影响[4]。通过识别过滤基因组数据中的敏感信息,可以实现基因组数据敏感信息脱敏,有助于降低基因组数据隐私敏感信息受攻击的风险。
基因组数据中的敏感序列包括STR 和DRS 这2 类。敏感序列识别串行算法的设计主要基于Hash映射思想,即通过产生序列Hash 指印来实现敏感序列的精确识别。此类代表算法有SRF(short read filter)[3]、LRF(long read filter)[4]和PriLive[5]等。这些基于序列Hash 指印思想的串行算法对高错误率的长序列基因组数据敏感序列识别准确率较低,且难以满足对大规模长序列实时识别过滤的要求。为此,一些学者基于多核CPU 集群系统,通过设计多核CPU 并行算法,在准确识别含有错误信息的敏感序列的同时,加速求解大规模基因组数据敏感序列识别问题。
关于基因组数据敏感序列识别并行算法的研究,文献[6]基于STR 挖掘工具FLASH(fast length adjustment of short read)和PERF(Python exhaustive repeat finder),在Hadoop 平台上开发了CPU 并行加速的基因组数据短串联重复序列识别算法BigFiRSt。文献[7]采用划分后缀分组方法,提出基于CPU 集群系统的最大串联重复序列识别并行算法。在串行算法FindTRs 的基础上,文献[8]设计了在多核CPU 集群系统上运行的大规模短串联重复序列识别并行算法MPI-dot2dot。根据STR 的周期重复性质,文献[9]在多核CPU 集群系统上设计实现了生物序列模式精确匹配的并行算法。文献[10]通过枚举所有具有统计显著性的局部最优出现频率的子串,提出了一种面向多核CPU 系统的频繁子串模式挖掘并行算法。上述多核CPU 并行算法仅针对基因组数据中的STR 进行识别,并没有处理基因组数据中DRS 的识别,因此这些并行算法仅能识别出基因组数据中的部分敏感序列。对于低错误率的基因组数据,文献[11]提出一种多核CPU 并行算法,以识别基因组数据中的短串联重复序列和疾病相关序列。
已有的基因组数据敏感序列识别并行算法的设计主要是针对敏感序列2 种类型(STR 和DRS)中的一种进行处理的;有的并行算法虽然同时处理STR 和DRS 的识别,但它面向的是低错误率序列构成的基因组数据,而不是高错误率序列构成的基因组数据。此外,已有的基因组数据敏感序列识别并行算法是多核CPU 并行算法,随着单分子实时测序和牛津纳米孔测序等第三代测序技术的发展,测序平台产生的序列具有长度长和错误率高的特点[12]。已有的并行算法难以高效、准确地识别出高错误率长序列构成的基因组数据中的2 类敏感序列STR 和DRS。为解决此问题,本文研究改进序列相似度计算模型,进而提出一种CPU 和GPU 协同计算加速识别高错误率长序列基因组数据中STR 和DRS 的并行算法CGPU-F3SR。该并行算法的特色和创新介绍如下。
1) 采用滑动窗口策略和k-mer 编码方法,分割并提取高错误率长序列基因组数据中的核苷酸序列碱基错误信息。引入布隆过滤器,对高错误率长序列基因组数据进行高效过滤,避免算法对重复数据的多次计算。
2) 根据短串联重复序列和疾病相关序列的结构特点,提出一种改进的序列相似度计算模型,实现准确识别出高错误率长序列基因组数据中的2 类敏感序列(STR 和DRS)。
3) 对基因组数据测序序列进行周期划分,设计实现CPU 和GPU 协同计算的敏感序列识别并行算法,大大提升算法的运行效率。
1 方法
设待识别基因组数据共有l条长序列,Ns[0:ns-1]表示长度为ns的第s条长序列,s=0,1,2,…,l-1。本文提出的CPU 和GPU 协同计算的高错误率长序列基因组数据敏感序列识别并行算法包括序列分割过滤、短串联重复序列并行识别、疾病相关序列并行识别和生成掩码核苷酸序列4 个阶段。
1.1 并行算法4 个阶段
1.1.1 序列分割过滤
为并行识别高错误率长序列基因组数据中的敏感序列,首先将长序列Ns[0:ns-1]分割为ns-r+1条、每条长度为r的短序列Rs,j=Ns[j:j+r-1],其中,短序列Rs,j和Rs,j+1共享r-1 个重叠碱基,j=0,1,2,…,ns-r,s=0,1,2,…,l-1,ns>r。因为人类DNA序列中约99.5%的序列具有高度一致性,所以将长序列分割为短序列会产生许多相同的短序列[3]。
布隆过滤器由一个二进制位数组(位向量)和若干Hash 函数组成,多用于数据去重以及数据检索[13]。为避免对重复短序列的多次计算,本文引入双布隆过滤机制对短序列Rs,j并行过滤去重,j=0,1,2,…,ns-r,s=0,1,2,…,l-1。
设布隆过滤器BF1用于敏感序列去重,其位数组和使用的Hash 函数数目分别为B1[0:b1-1]和h1。布隆过滤器BF2用于非敏感序列去重,其位数组和使用的Hash 函数数目分别为B2[0:b2-1]和h2。为了初始化双布隆过滤器,需根据式(1)计算B1和B2的位数组规模b1和b2[13]。
其中,pi表示布隆过滤器误报率,ti表示BFi可容纳序列数目的上限。通过预设参数pi和ti,可以根据式(1)计算得到位数组规模bi,i=1,2。此时,可以计算BFi使用的Hash 函数数目hi[13]为
依据式(1)和式(2)计算出双布隆过滤器的参数b1和b2以及h1和h2之后,初始化布隆过滤器位数组B1,i=0 和B2,k=0,i=0,1,2,…,b1-1,k=0,1,2,…,b2-1。
当初始化双布隆过滤器完成后,将短序列Rs,j进行周期划分,然后使用Pr 个CPU 核心通过双布隆过滤器BF1和BF2并行地对短序列Rs,j进行过滤去重,s=0,1,2,…,l-1,j=0,1,2,…,ns-r。
为方便理解短序列周期划分思想,图1 给出了块划分和周期划分短序列的示例。
人类基因组数据测序序列中存在连续的串联重复区域。若将分割得到的短序列采用图1(a)所示块划分方式并行处理,则会造成部分线程处于“计算等待”状态。为均衡数据计算,使每个线程达到负载均衡,本文采用图1(b)所示的周期划分对短序列进行并行处理,并采用双布隆过滤器过滤去重。
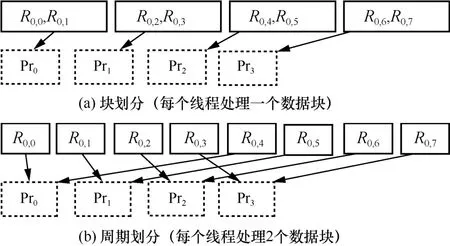
图1 基于块划分和基于周期划分短序列的示例
本文并行短序列过滤去重方法介绍如下。
Pr 个CPU 核心并行对Rs,v+dPr执行h1次Hash计算,以获得h1个Hash 值H1,i(Rs,v+dPr),且对BF1中的位数组B1进行查询。
若B1[H1,i(Rs,v+dPr)]的值均为1,i=0,1,2,…,h1-1,则表明Rs,v+dPr是重复短序列且为敏感短序列;若B1[H1,i(Rs,v+dPr)]的值不全为1,则需对Rs,v+dPr执行h2次Hash 计算,以获得h2个Hash 值H2,i(Rs,v+dPr),并对BF2中的位数组B2进行查询。
若B2[H2,i(Rs,v+dPr)]的值均为1,i=0,1,2,…,h2-1,则表明Rs,v+dPr为重复短序列且为非敏感短序列;若B2[H2,i(Rs,v+dPr)]的值不全为1,则表明Rs,v+dPr不存在于BF1或BF2中,Rs,v+dPr不是一条重复序列,此时需对Rs,v+dPr进行短串联重复和疾病相关序列识别处理,d=0,1,2,…,,v+dPr≤ns-r,v=0,1,2,…,Pr-1,s=0,1,2,…,l-1。
通过并行地对短序列过滤去重,可避免对短序列的重复计算。若短序列不存在于布隆过滤器BF1或BF2,则需对短序列进行短串联重复序列识别的处理。
1.1.2 短串联重复序列并行识别
STR 也被称作微卫星DNA,由1~6 个碱基的重复基序组成,约占人类基因组数据的3%[3]。短串联重复序列的重复结构性质表明,通过合理分割短串联重复序列,可以得到记录短串联重复序列基本重复单元的相同子段[14]。在借鉴文献[14]思想的基础上,本文使用Pr 个CPU 核心并行地识别高错误率长序列基因组数据中的短串联重复序列。
并行识别短串联重复序列的方法介绍如下。
首先,Pr 个CPU 核心并行分割短序列Rs,v+dPr为4 个长度均为w的短片段集合i=0,1,2,3,v=0,1,2,…,Pr-1,v+dPr≤ns-r,s=0,1,…,l-1。
为降低将非敏感序列误识别为短串联重复序列的概率,根据短串联重复序列的结构特点,文献[15]引入控制参数c_str 和皮尔逊相关系数(PCC,Pearson correlation coefficient)改进序列局部片段相似度的计算为
1.1.3 疾病相关序列并行识别
DRS 是指携带疾病易感基因的核苷酸序列[11]。不同于短串联重复序列,疾病相关序列更多体现在位点上的变异,而非结构上的重复。为此,本文通过改进序列相似度计算模型,将待识别序列和第三方数据库中疾病相关序列进行计算比对,以并行识别出高错误率长序列基因组数据中的疾病相关序列。
1) 并行构建敏感序列词典
构建敏感序列词典是为了降低大量短序列与第三方数据库中疾病相关序列相似度计算的代价。本文使用二维数组 hostDict[0:m-1][0:4k+2]表示CPU 端构建包含m条序列的敏感序列词典。
并行构建敏感序列词典的方法介绍如下。
首先,Pr 个CPU 核心并行地从第三方数据库提取每个疾病易感基因的染色体编号CHR_ID 和染色体位置CHR_POS,并使用pysam 工具[16]从人类参考基因组HG38 中提取m条长度为r的疾病相关序列Dv,i,i=0,1,2,…,m-1,v=0,1,2,…,Pr-1。
2) 疾病相关序列相似度并行计算
在判断短序列Rs,j是否为疾病相关序列之前,本文已经对Rs,j进行过滤去重和短串联重复序列识别。因此,仅需要对经上述步骤处理后剩余的g条序列进行疾病相关序列识别。将剩余的g条序列和敏感序列词典hostDict[0:m-1][0:4k+2]中的m条序列进行相似度计算比对十分耗时。为此,本文采用多核CPU 和GPU 协同并行计算以加速疾病相关序列的识别,j=0,1,2,…,ns-r,s=0,1,2,…,l-1。
CUDA(computer unified device architecture)并行编程模型将GPU 线程层次结构抽象成线程块网格Grid 和线程块Block 两层[17]。在CUDA 线程层次结构中,Block 和Grid 由uint3 类型定义的三维向量 blockIdx.x、blockIdx.y、blockIdx.z 和threadIdx.x、threadIdx.y、threadIdx.z 表示[17]。为准确定位识别疾病相关序列,本文采用一维线程块网格和线程块进行CUDA 线程索引。
其中,e=0,1,2,i=0,1,…,m-1,tID=0,1,…,g-1,s=0,1,…,l-1,dis_sim 为疾病相关序列相似度阈值。若sim(Di,Rs,tID)≥dis_sim,则序列Rs,tID是一条疾病相关序列,且它和敏感序列词典中第i条序列Di具有很高的序列相似度。
1.1.4 生成掩码核苷酸序列
1.2 并行算法描述与分析
设GWAS Catlog 数据库中疾病相关序列数据集为disData(包含m组数据),HG 表示疾病相关序列并行识别步骤构建敏感序列词典时所使用的参考基因组,并行算法使用的CPU 核心数为Pr,使用的GPU 线程数为Cu。
图2 给出了CPU/GPU 并行算法CGPU-F3SR的处理流程。
图2 中,res[0:ns-1]表示记录每条短序列的敏感序列识别结果的辅助数组,且res[j]∈{0,1,2,3,4}分别表示原始短序列结果标记、BF1过滤标记、BF2过滤标记、短串联重复序列标记和疾病相关序列标记,j=0,1,2,…,ns-1,s=0,1,2,…,l-1。hostRes[0:g-1]表示保存疾病相关序列的识别结果的辅助数组,其中,,I(res[j])=0 表示指示函数(条件成立则取值为1,否则取值为0)。strIndex[0:g-1]表示保存疾病相关序列识别的短序列索引的辅助数组。式(4)中,的求解需要做许多分支判断。若使用GPU 计算,则将使部分GPU 线程处于长时间“计算等待”状态,为均衡 GPU 端数据运算,本文引入辅助数组hostData[0:g-1][0:4k+2],并使用多核CPU 预编码核苷酸序列和碱基含量统计,以简化GPU 端数据运算,提升GPU 端的运行效率。
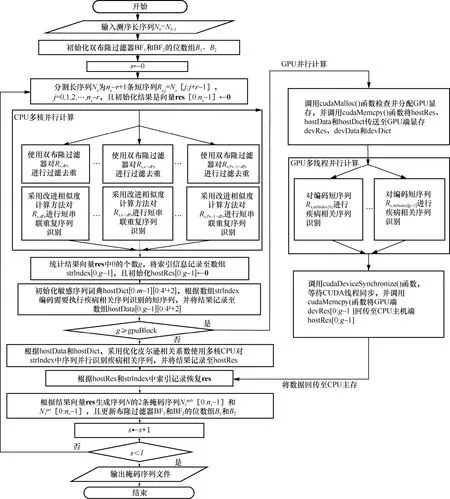
图2 CPU/GPU 并行算法CGPU-F3SR 的处理流程
算法1 给出了CGPU-F3SR 的流程。
算法1 CGPU-F3SR




当CPU/GPU 并行算法CGPU-F3SR 中参数的gpuBlock 取值为0 时,它就变为仅采用多核CPU并行计算的版本(简记为算法CPU-F3SR)。
2 实验
实验在广西大学的Sugon 7000A 超级并行计算机上进行,使用CPU/GPU 异构计算节点。每个计算节点的内存容量为512 GB、外部存储容量为8×900 GB、CPU 为2×Intel Xeon Gold 6230(总内核数为40)、GPU 为NVIDIA Tesla-T4(显存容量为16 GB,含有2 560 个CUDA 核心)。运行操作系统为64 位版本CentOS7.4。采用C++、CUDA 和OpenMP 混合编程实现并行算法CGPU-F3SR。
实验采用GWAS Catalog 数据库[18]中全基因组关联研究数据集gwas_catalog_v1.0.tsv构建敏感序列词典,该数据集包含Scoliosis、HIV-1 replication 和Opioid sensitivity 等4 680 种疾病,共计216 250 组数据。采用的参考基因组为NCBI 数据库[19]开放的人类基因组HG38。实验所用的疾病易感基因来自GWAS Catalog 数据库[18],短串联重复序列来自TRDB 数据库[20]。参考文献[3]的数据预处理规则,预处理每条序列长度为50 bp 的基准数据序列集All-Together(2 239 340 条序列),每个基准数据集的非敏感序列和敏感序列数量之比为7:1。
为评估算法识别效果,需获取不同长度且带有敏感序列标签的测序长序列。为此,实验对错误率为2%~20%的All-Together 基准数序列集进行随机抽样,生成序列长度为100~400 kbp 的总规模约24 GB 的4 组长序列数据集DataSet-100 kbp~Data Set-400 kbp 用于并行算法参数调优测试。
实验将本文CPU 和GPU 协同计算的并行算法CGPU-F3SR与并行算法PARA-LRF[11]、仅采用CPU并行计算的版本CPU-F3SR(gpuBlock=0)、串行算法F3SR[15]进行测试比较识别效果。实验评估算法的识别准确率(acc,accuracy)[3]、查准率(pre,precision)[4]和假阳性率(fpr,false positive rate)[4]。准确率和查准率越高,算法识别效果越好;假阳性率越低,算法识别效果越好。在运行效率方面,实验评估了算法的识别吞吐量(thp,throughput)[4]。吞吐量定义为算法单位时间识别脱氧核糖核苷酸的数量,单位为nt/s[3-4],吞吐量越高,算法运行效率越高。
下面,首先给出本文所提出的CPU/GPU 并行算法CGPU-F3SR 的参数调优实验结果,然后给出消融实验结果,最后给出算法CGPU-F3SR 和同类并行算法的实验结果及分析。
2.1 并行算法参数调优实验
参考文献[3,4,15]给出的基因组数据敏感序列识别算法研究的相关实验,算法CGPU-F3SR 选取pi=0.000 1、ti=108、r=48 bp、k=5、str_sim=0.64、dis_sim=0.80 进行算法部分参数的初始化,i=1,2。
为确定GPU 线程块大小对并行算法吞吐量的影响,实验测试了并行算法CGPU-F3SR 采用不同的GPU 线程块大小在4 组长序列数据集上运行时的吞吐量,结果如图3 所示。

图3 并行算法CGPU-F3SR 采用不同的GPU 线程块大小在4 组长序列数据集上运行时的吞吐量
图3 实验结果表明,当GPU 线程块大小固定时,序列数据集规模越大,并行算法CGPU-F3SR 的吞吐量越高。原因如下:1) 测序长序列数据集规模越大,CGPU-F3SR 分割长序列得到的短序列中的重复短序列越多,由于双布隆过滤避免了重复短序列的多次运算,因此过滤效果明显;2) CGPU-F3SR 将编码后的短序列提交给GPU 进行疾病相关序列并行识别,测序数据集越大,GPU 线程平均计算处理的数据越多,算法的并行度越高。按照CUDA 并行模型,网格Grid中的各线程块Block 会被分配到GPU 的各流式处理器中执行。图3 的实验结果还表明,当参数gpuBlock取值为128~384 时,算法整体上具有较高的吞吐量。
图4 进一步给出了GPU 线程块大小gpuBlock=256、使用不同 CPU 核心数运行并行算法CGPU-F3SR 时得到的吞吐量结果。
图4 实验结果表明,并行算法CGPU-F3SR 的识别吞吐量与使用的CPU 核心数呈正相关。数据集规模越大,序列越长,算法吞吐量受CPU 核心数增益效果越明显。
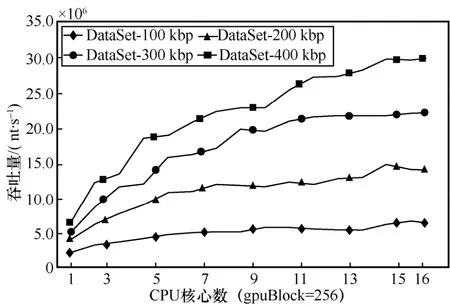
图4 并行算法CGPU-F3SR 使用不同CPU 核心数在4 组长序列数据集上运行时的吞吐量
为使本文并行算法CGPU-F3SR 充分发挥CPU和GPU 协同并行计算的能力,根据图3 和图4 的实验结果,本文选取gpuBlock=256 和Pr=4 的组合进行后续的实验。
2.2 并行算法消融实验
并行算法CGPU-F3SR 引入布隆过滤器对序列数据进行过滤,以免对分割短序列的重复计算。为了确定过滤机制对并行算法CGPU-F3SR 的性能影响,本文实验评估了并行算法CGPU-F3SR 进行序列过滤With Filter(参数Pr=4,gpuBlock=256)和不进行序列过滤 Without Filter(参数 Pr=30,gpuBlock=256)2 种情形在4 组长序列数据集运行得到的识别准确率、查准率、假阳性率和吞吐量,实验结果如表1 所示。
表1 的消融实验结果表明,在识别性能方面,并行算法CGPU-F3SR 在With Filter 和Without Filter 这2 种情形下均获得相同的识别准确率、查准率和假阳性率。这是因为:1) 布隆过滤器的多哈希映射机制可以对相同序列产生多个相同的哈希映射,这可以在避免对重复序列的多次计算的同时不影响其识别性能;2) 改进序列相似度计算模型能对含有错误信息的相似序列产生近似度量值,并行算法CGPU-F3SR 的识别准确率、查准率和假阳性率均取决于改进的序列相似度计算模型,而不是取决于布隆过滤器。在运行效率方面,算法使用4 个CPU核心的With Filter 模式的识别吞吐量比使用30 个CPU 核心的Without Filter 模式高了2 个数量级。这是因为长序列基因组数据中分割短序列具有大量相同的重复短序列,布隆过滤效果明显。

表1 并行算法CGPU-F3SR 的识别准确率、查准率、假阳性率和吞吐量
2.3 并行算法识别性能实验
为评估算法的并行化对高错误率长序列基因数据中敏感序列(包括STR 和DRS)识别效果的影响,首先,实验测试CPU 和GPU 协同并行算法CGPU-F3SR、仅CPU 并行的算法CPU-F3SR 和串行算法F3SR(参数取值pi=0.000 1,ti=104,r=48,k=5,dis_sim=0.80,str_sim=0.64,i=1,2)[15]在4 组长序列数据集上运行得到的识别准确率、查准率和假阳性率,实验结果如表2 所示。
表2 的实验结果表明,CPU 与GPU 协同并行算法CGPU-F3SR 和CPU 并行化算法CPU-F3SR 均获得了与串行算法F3SR 相同的识别结果。这是因为长序列分割得到的相邻短序列之间共享r-1 个高度重叠的碱基,通过CPU/GPU 并行计算不会影响算法的最终识别质量。在上述实验的基础上,进一步测试比较本文并行算法CGPU-F3SR 和同类并行算法PARA-LRF(参数p=0.02,m=68 915 861)[11]在DataSet-100 kbp~DataSet-400 kbp 数据集上运行时,对高错误率长序列基因组数据中敏感序列(包括STR 和DRS)进行识别获得的识别准确率、查准率和假阳性率,实验结果如表3 所示。
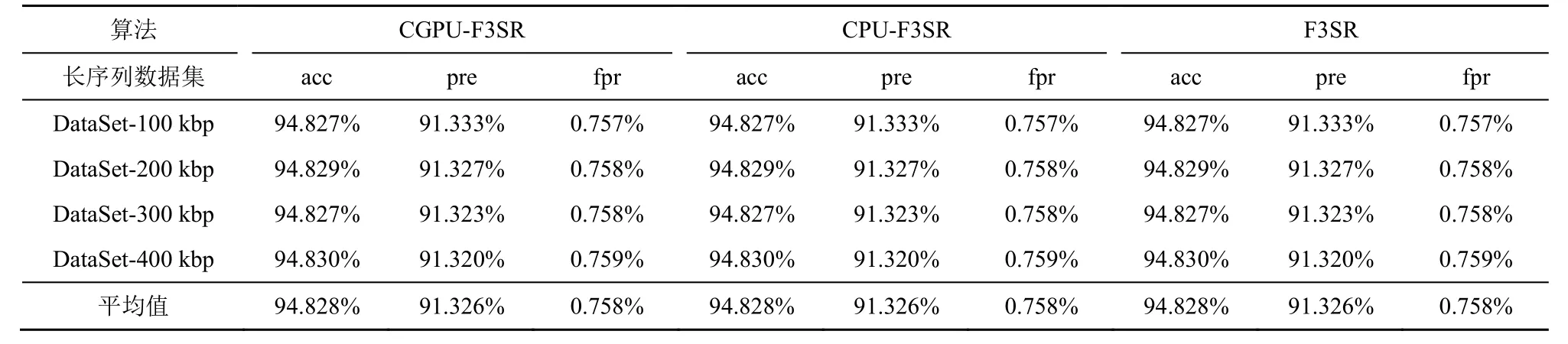
表2 并行算法CGPU-F3SR、CPU-F3SR 和F3SR 的识别准确率、查准率和假阳性率
表3 的实验结果表明,相较于并行算法PARA-LRF,本文CPU/GPU 并行算法CGPU-F3SR整体上具有更高的识别准确率和查准率以及更低的识别假阳性率。这是因为并行算法CGPU-F3SR采用k-mer 编码策略有效地提取了高错误率长序列中的碱基错误信息,且根据STR 和DRS 的结构特点,通过引入控制参数改进了STR 和DRS 相似度的计算方法,使本文并行算法CGPU-F3SR 在准确识别出高错误率长序列基因组数据中敏感序列的同时,降低了将非敏感序列误判为敏感序列的情形的发生,从而整体上获得更好的识别效果。

表3 并行算法CGPU-F3SR 和PARA-LRF 的识别准确率、查准率和假阳性率
2.4 并行算法运行效率实验
在基因组数据敏感序列识别研究中,算法吞吐量和运行时间相关,多用于运行效率评估。基因组数据敏感序列识别并行算法相关研究较少,目前缺乏CPU 和 GPU 协同计算的并行算法。并行算法PARA-LRF[11]是基于多核CPU 设计的并行算法。为公平起见,实验首先测试了本文仅多核CPU 并行算法CPU-F3SR和多核CPU并行算法PARA-LRF[11]使用不同CPU核心数在4组长序列数据集上运行得到的识别吞吐量,结果如图5 所示。
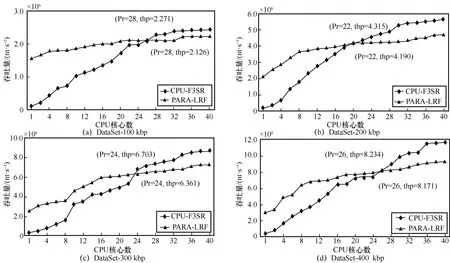
图5 多核CPU 并行算法CPU-F3SR 和PARA-LRF 使用的CPU 核心数对吞吐量的影响
从图5(a)可以看出,在DataSet-100 kbp 数据集上,当使用的CPU 核心数为1~28 时,CPU 并行算法PARA-LRF 的识别吞吐量高于本文CPU 并行算法CPU-F3SR;当使用的CPU 核心数为28~40时,并行算法CPU-F3SR 比PARA-LRF 获得了整体上更高的识别吞吐量。
从图5(b)~图5(d)可以看出,对序列长度为200~400 kbp 的3 个长序列数据集,当CPU 核心数分别为22、24 和26 时,并行算法CPU-F3SR 达到吞吐量拐点,此时本文多核 CPU 并行算法CPU-F3SR 比多核CPU 并行算法PARA-LRF 整体上具有更高的识别吞吐量。这是因为并行算法CPU-F3SR 并行过滤去重分割得到的短序列、并行识别短串联重复序列、并行识别疾病相关序列的部分占比高、算法并行度高,所以在4 组长序列数据集上运行时获得了更高的识别吞吐量。
从长序列基因组数据中识别疾病相关序列的过程是一个计算密集型工作,为进一步加速求解敏感序列识别问题,本文设计实现了CPU 和GPU 协同计算的敏感序列识别并行算法CGPU-F3SR。为此,实验也测试了 CPU/GPU 并行算法CGPU-F3SR(参数Pr=4、gpuBlock=256)、多核CPU并行算法PARA-LRF(参数p=0.02、m=68 915 861、Pr=4)[11]、多核CPU 并行算法CPU-F3SR(参数Pr=4)在4 组长序列数据集上运行得到的识别吞吐量,结果如表4 所示。
从表4 可以看出,相较于本文仅采用多核CPU并行加速的CPU-F3SR 算法,本文的CPU 和GPU协同并行求解敏感序列识别问题的CGPU-F3SR 算法平均吞吐量高达11.033×106nt/s,极大提升了识别吞吐量;相较于已有的多核 CPU 并行算法PARA-LRF,本文算法CGPU-F3SR 在吞吐量方面也具有较大幅度的提升。CGPU-F3SR 算法的平均吞吐量分别是CPU-F3SR 和PARA-LRF 算法的11.45 倍和2.44 倍。CGPU-F3SR 算法之所以获得更高的吞吐量,是因为它采用GPU 并行加速处理属于计算密集型的疾病相关序列识别过程,加速效果显著。
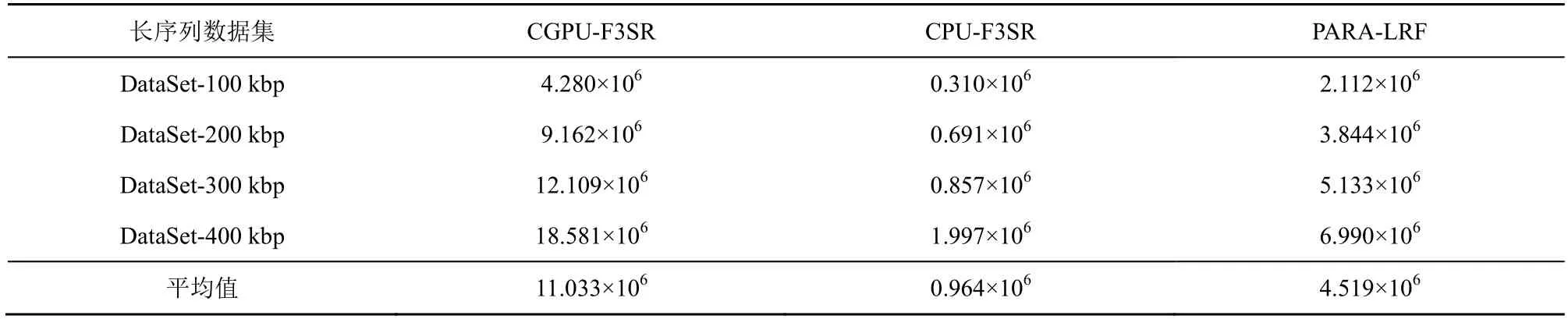
表4 并行算法CGPU-F3SR、CPU-F3SR 和PARA-LRF 的识别吞吐量
综上所述,CPU 和GPU 协同计算的并行算法CGPU-F3SR 在准确识别出大规模高错误率长序列基因组数据中的2 类敏感序列(STR 和DRS)的同时,显著提升了吞吐量。
3 结束语
第三代测序技术产生的基因组测序数据正以超摩尔定律的速度显著增长。随着人们生物信息安全意识的逐渐增强,保护个体基因组数据敏感序列变得越来越重要。本文提出的CPU 和GPU 协同计算识别的并行算法CGPU-F3SR 的特色和优势是高效准确地识别出第三代测序技术产生的大规模的高错误率长序列基因组数据中的2 类敏感序列(短串联重复序列和疾病相关序列)。在长度为100~400 kbp 的长序列数据集上的实验结果表明,并行算法CGPU-F3SR 在整体上获得了较高的识别准确率、查准率和较低假阳性率的同时,显著提升了识别吞吐量。
近些年来,学者研究发现基因组测序数据中还存在“均聚物”错误。考虑到基因组数据错误类型复杂、数据规模巨大、数据敏感的特点,在本文研究工作基础上,下一步的研究方向将包括:1) 研究设计能准确识别出“均聚物”错误的敏感序列识别算法;2) 探索设计新的数据索引结构或借鉴其他索引数据结构(如简洁de Bruign图结构、FM-index 结构等),研究设计在CPU/GPU 混合体系结构集群上实现的并行算法,以进一步加速求解超大规模超长序列的基因组数据敏感序列识别问题;3)研究如何将本文提出的敏感序列识别并行算法与安全比对算法进行级联,以实现大规模的高错误率长序列基因组数据的安全比对。
猜你喜欢
中国卫生统计(2023年5期)2023-11-30 01:40:14
科技创新导报(2021年31期)2021-05-10 14:55:00
教师·中(2017年3期)2017-04-20 21:49:49
环球市场(2017年36期)2017-03-09 15:48:21
湖南城市学院学报(自然科学版)(2016年2期)2016-12-01 04:06:43
试题与研究·教学论坛(2016年27期)2016-08-11 14:57:08
教学研究与管理(2014年4期)2014-05-16 22:44:12
电子设计工程(2014年18期)2014-02-27 12:00:14
吉林建筑大学学报(2012年3期)2012-08-15 00:54:52
杭州电子科技大学学报(自然科学版)(2010年5期)2010-01-08 07:28:38
