
陆地棉高密度遗传图谱的构建及产量相关性状的QTL定位
2023-03-09 08:40:08贾晓昀王士杰朱继杰赵红霞李妙王国印
中国农业科学 2023年4期
贾晓昀,王士杰,朱继杰,赵红霞,李妙,王国印
河北省农林科学院粮油作物研究所/河北省作物遗传育种实验室/河北省作物栽培生理与绿色生产重点实验室,石家庄 050035
0 引言
【研究意义】中国是世界上棉花消费量最大的国家。据中国棉花协会官方统计,近年来,中国的棉花产量始终无法满足消费量,存在较大缺口,年进口量居高不下(https://www.china-cotton.org/data/demandData)。此外,由于当前国际环境的波动性增加,棉花市场的稳定性较差。因此,保障中国棉花的自给能力对国内纺织工业的发展具有重要现实意义,高产育种仍然是目前棉花育种的主要任务[1]。【前人研究进展】衣分、子指和单铃重是棉花产量的重要构成性状,且均为典型的数量性状,QTL定位是挖掘其调控基因的主要方法之一[2]。自1994年第一张棉花遗传图谱公布以来[3],棉花重要性状的分子遗传基础研究拉开序幕,大量基于以SSR标记为代表构建的遗传图谱和产量相关QTL位点被公布[4]。但是,由于对陆地棉基因组信息的了解较少,加之标记密度较低,标记开发及材料的基因型鉴定过程费时费力,前期QTL定位结果的准确度和精度普遍偏低。二倍体雷蒙德氏棉的参考基因组首先于 2012年公布[5-6],随后二倍体的亚洲棉[7]、异源四倍体的陆地棉[8-9]等棉种的参考基因组陆续公布,并且陆地棉TM-1参考基因组的组装质量不断提高[10-11],NDM8参考基因组的公布进一步促进了对中国自育优异品种基因组结构和变异的认识[12]。基于此,曲朝阳等[13]、WANG 等[14]利用一张包含6 295个SNP的遗传图谱,分别定位到32个单铃重 QTL和 28个衣分 QTL。ZHANG等[15]利用SLAF-seq技术构建了一张包含5 521个SNP的高密度遗传图谱,在11个环境下定位到18个稳定的单铃重QTL,注释到344个候选基因。利用高质量的整合图谱,ZHANG等[2]在17个环境下进行棉花产量和纤维品质的QTL定位,综合分析了产量和纤维品质的分子遗传基础和相互关系。DIOUF等[16]利用GBS技术构建了一张包含5 178个SNP的高密度遗传图谱,定位到13个棉花产量QTL。GU等[17]构建了一张包含6 187个bin的高密度遗传图谱,定位60个产量QTL,预测了1个衣分的候选基因。然而,目前,基于高密度SNP遗传图谱开展棉花产量性状的 QTL定位研究仍然较少,已知的产量候选基因暂时无法充分阐释相关性状的分子调控机理。【本研究切入点】目前,限制分子标记辅助育种效率的主要原因是对棉花产量相关的调控基因或分子标记了解较少,而且,用于棉花产量性状QTL定位的高密度SNP遗传图谱数量较少,不同材料间QTL定位结果的重复性较差,优异基因挖掘的效率较低。冀丰1271由河北省农林科学院粮油作物研究所选育,株型舒朗通透,具有突出的高产、稳产、抗烂铃等特征,为目前河北省区域试验对照品种。冀丰173为河北省农林科学院粮油作物研究所选育的自交系,具有纤维品质优异、结铃吐絮集中等特征。二者在产量性状方面差异极显著。【拟解决的关键问题】本研究以冀丰1271为母本、冀丰173为父本构建一个新的遗传分离群体,利用简化基因组测序技术构建高密度SNP遗传图谱,开展多群体产量相关性状的QTL定位,获得稳定性好、精确度高的QTL,为产量性状调控基因的挖掘和有效分子标记的开发提供更多研究基础。
1 材料与方法
1.1 试验材料与设计
以冀丰1271(冀审棉2012002号)为母本、冀丰173(自交提纯)为父本,于2018年夏季在河北省农林科学院粮油作物研究所堤上试验站(石家庄)完成杂交组配,同年冬季在河北省农林科学院粮油作物研究所南繁基地(三亚南红农场)加代种植F1并自交。于2019年夏季在石家庄种植F2群体(402个单株),行长7 m,行距0.7 m,株距0.2 m。根据编号顺序,从F2群体中选择前200个正常生长的单株连续自交,于2020年和2021年夏季在石家庄分别种植F2:3和F2:4群体,行长4 m,行距0.7 m,株距0.2 m,随机区组设计,2次重复。常规大田管理。
1.2 性状调查与数据分析
调查衣分(lint percentage,LP)、子指(seed index,SI)和单铃重(boll weight,BW)3个产量性状。自然吐絮后,人工采收,F2群体单株收获并计数,F2:3和F2:4群体每行收获植株中部30铃,称重计算单铃重,轧花后称取皮棉重量,计算皮棉与籽棉重量的比值即为衣分,数出100粒正常的种子称重即为子指。
F2群体的单株性状即为表型性状,F2:3和F2:4群体重复的平均数为表型性状。采用Excel 2010计算基本统计量,采用SPSS17计算性状之间的相关性。
1.3 简化基因组测序与构建遗传图谱
采用CTAB法提取F2群体幼叶DNA,检测合格后,利用序基因分型(genotyping by sequencing,GBS)[18-19]方法开发SNP。以MseⅠ和TaqαⅠ(Thermo scientific Fermentas)2种酶对DNA进行酶切;链接接头;胶回收397—420 bp的DNA片段(Qiagen,Valencia,CA),并进行 PCR扩增(Phusion High-fidelity,Finnzymes)后,在Illumina HiSeqTM平台进行测序。根据LI等[20]数据分析方法过滤下机数据,并以BWA(0.7.17)软件[21]将高质量序列比对至TM-1参考基因组[10]。利用GATK(4.0.11.0)软件[22]鉴定SNP标记。利用MSTMap软件[23]构建遗传图谱。
1.4 QTL定位与基因分析
采用 WinQTLCart 2.5软件复合区间作图法(composite interval mapping,CIM)进行QTL定位,窗口大小为5 cM,步长为1 cM,背景标记为10个,LOD值为2.5。贡献率在10%以上的QTL为主效QTL,至少在2个群体内检测到的QTL为稳定QTL。根据参考基因组信息,注释主效和稳定QTL的95%置信区间内的基因,利用在线工具 KOBAS(http://kobas.cbi.pku.edu.cn/)进行KEGG和GO分析,利用TM-1[24]和NDM8[12]的转录组数据分析基因的表达模式。
2 结果
2.1 测序数据质量统计
由表1的统计数据可知,经过对GBS下机数据过滤,母本冀丰 1271得到 26.93 Gb数据,Q30值为90.55%,父本冀丰173得到27.30 Gb数据,Q30值为89.95%,200个F2单株共得到328.84 Gb数据,平均Q30值为95.77%,均达到测序深度要求的数据量和数据质量。经过与参考基因组比对,冀丰 1271和冀丰173的比对率分别为99.64%和99.47%,测序深度分别为9.58×和9.69×,对基因组的覆盖度分别为86.45%和 84.28%;F2群体的比对率为 99.69%,平均深度为0.70×,覆盖度为 4.17%。测序数据已上传至 NCBI数据库(PRJNA867015)。

表1 过滤后的测序数据量、数据质量及比对至参考基因组的基本统计量Table 1 Number and quality of the clean reads and statistics of mapping with the reference genome sequence
以F2群体为试验材料开发分子标记,共得到1 305 642个SNP,分为8类标记类型,其中,用于后续遗传图谱构建的aa×bb型SNP标记有410 726个(表2)。

表2 标记类型及数量统计Table 2 Statistics of marker type and marker number
2.2 遗传图谱信息
首先,根据标记在参考基因组上的位置,将标记锚定到26条染色体。其次,根据标记在F2群体中的交换重组关系,计算标记之间的顺序和相对距离,在LOD值为4—20,逐条染色体构建连锁群(表3),最终上图的SNP标记为16 088个,其中SNP最少的是D3染色体(39),最多的是D5染色体(1 746),说明标记在各条染色体分布的均匀性较差,可能是因为亲本间遗传多样性较低。图谱总长度为4 282.81 cM,标记间的平均距离为0.27 cM,最大的Gap为D3染色体上的19.75 cM。由图1标记共线性及其相关系数分析可知,上图标记的顺序与其物理位置的顺序一致性较好,证明构建的高密度遗传图谱质量较高。
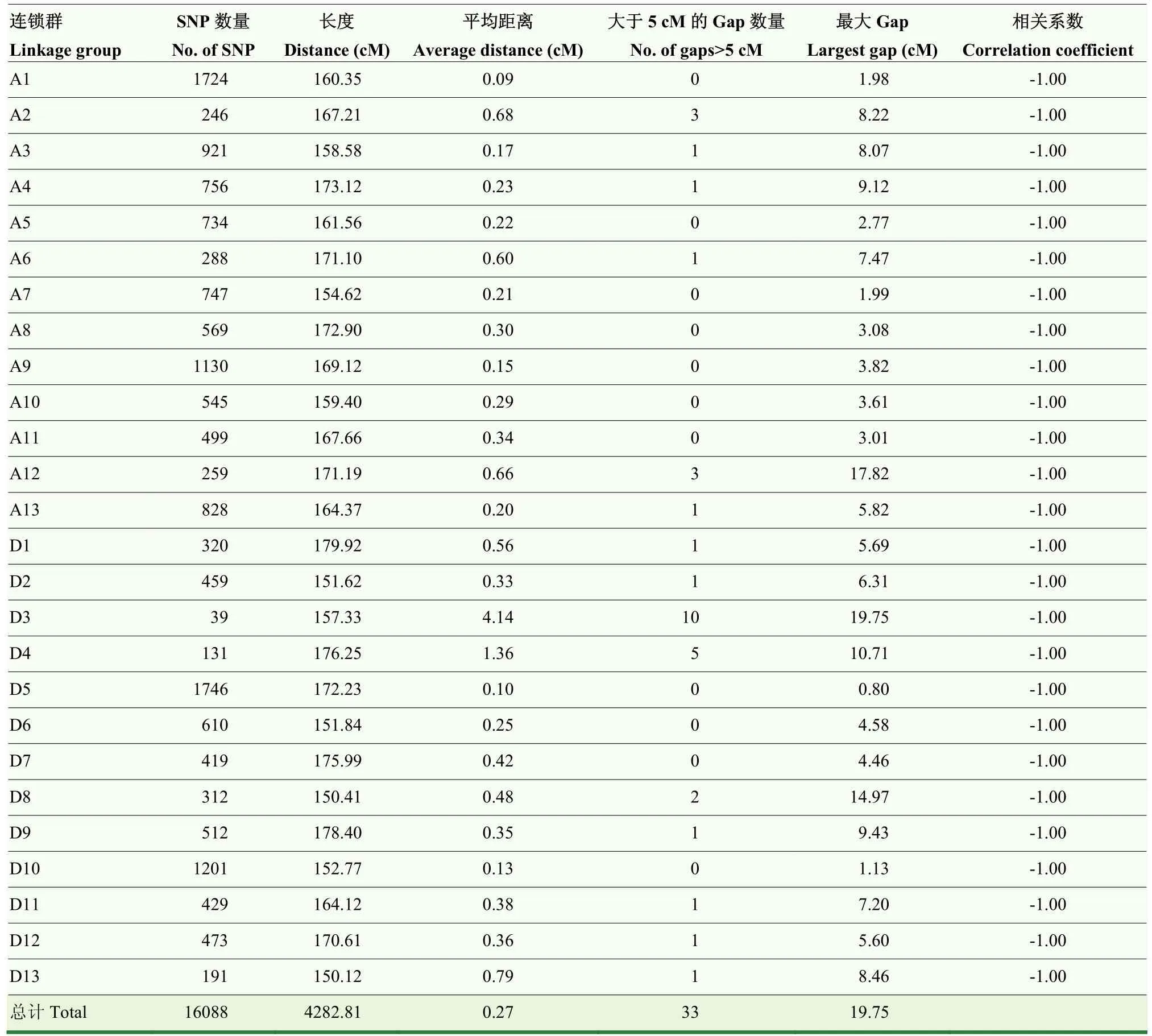
表3 各条连锁群的详细信息Table 3 Detail information of each linkage group

图1 上图标记与其参考基因组物理位置的共线性分析Fig.1 Collinearity analysis of the mapped markers on the genetic map with their physical position on the reference genome
2.3 表型数据分析
由表4可知,冀丰1271的衣分、子指和单铃重均显著或极显著高于冀丰173,说明2个亲本材料的产量性状差异较大,可用于构建分离群体开展QTL定位研究。F2、F2:3和F2:4群体的衣分、子指和单铃重均表现为双向超亲分布,峰度和偏度的绝对值均小于 1,说明衣分、子指和单铃重在分离群体中呈近似正态分布,冀丰1271和冀丰173中均含有调控3个目标性状的微效基因。由变异系数可知,单铃重的变异系数最大(6.87%—16.24%),衣分的变异系数最小(3.03%—3.73%)。由表 5相关性分析结果可知,衣分与子指呈极显著负相关,子指与单铃重呈极显著正相关,而衣分与单铃重呈不显著的正相关关系。
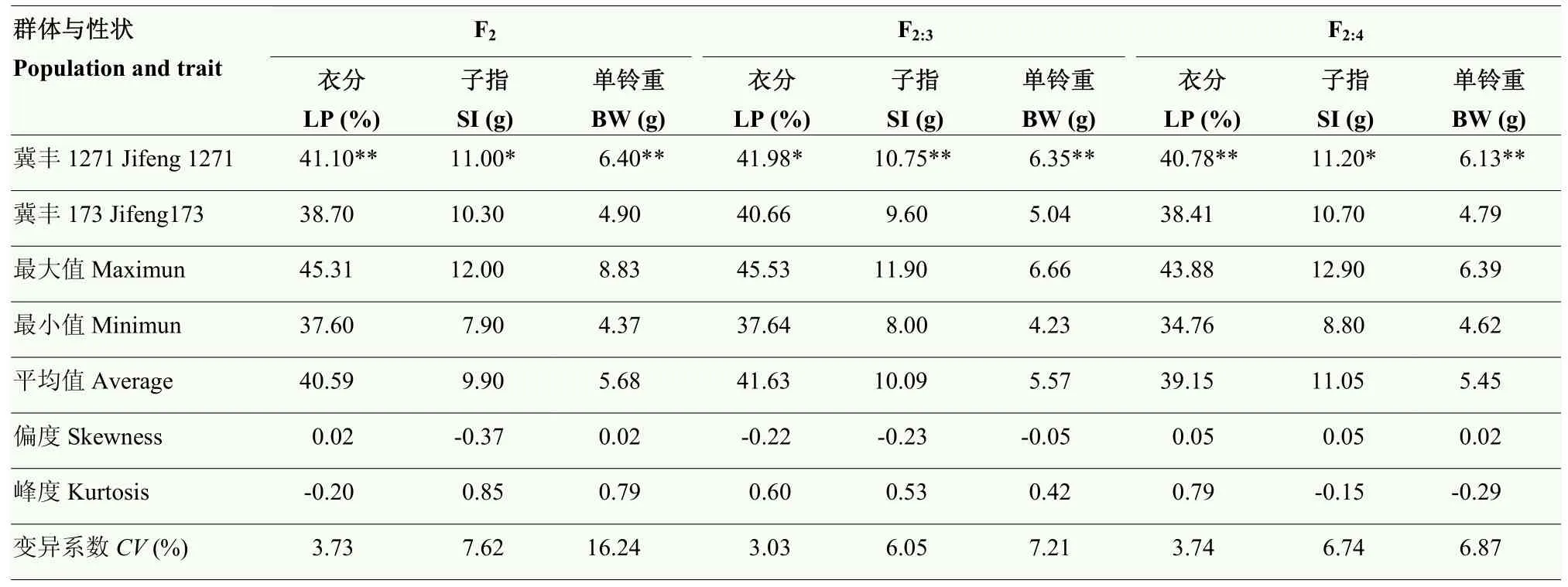
表4 亲本及群体的表型数据统计量Table 4 Phenotypic statistics of the parents and populations

表5 衣分、子指和单铃重的相关性分析Table 5 Correlation analysis of boll weight, lint percentage and seed index
2.4 QTL定位结果
在3个群体中共定位到108个QTL(电子附表1),包括34个衣分QTL、36个子指QTL和38个单铃重QTL。至少在 2个群体中被检测到的稳定 QTL共有16个,包括9个衣分QTL、4个子指QTL和3个单铃重QTL。单个 QTL的贡献率为 1.07%—24.72%,贡献率大于10%的主效QTL共有10个,包括4个衣分QTL、2个子指QTL和4个单铃重QTL。QTL分布于23条染色体,各染色体上的QTL数量为1—12个(A6、D3和D6染色体均为0),分别有51和57个QTL定位于A和D基因组。有57个QTL的增效基因来源于冀丰1271,47个QTL的增效基因来源于冀丰173,4个稳定QTL的增效基因在不同群体中的来源不同。发现一个衣分QTL(qLP-A13-4)可以在3个群体中定位到,贡献率为6.54%—13.78%,增效基因来源于冀丰1271。
2.5 基因注释与筛选
利用基因组注释信息,在 21个主效和稳定的QTL内共注释到3 415个基因,通过与GO和KEGG数据库比对,富集到的前20个GO条目主要包括细胞质、内质网、高尔基体等细胞组分,信号受体活性、蛋白酶体结合、脱落酸结合等分子功能,以及磷饥饿的细胞响应、脱落酸激活的信号途径等生物过程(图2);富集到的前20个KEGG条目主要包括植物激素信号转导、TCA循环、光合生物的碳固定以及物质的合成与代谢等通路(图3)。根据KEGG比对结果发现,在前20个比对到的通路中共有279个基因,主要参与信号转导、物质合成与代谢,可能包含有参与棉花产量形成的关键调控基因。通过分析基因在TM-1[24]和NDM8[12]中的表达,发现8个基因在不同组织中具有较高的表达量(电子附表2)。Ghir_A13G010390、Ghir_D02G015800和Ghir_D13G010980在 TM-1的胚珠和纤维中高调表达,Ghir_D10G018940和Ghir_D13G005390在TM-1的0和1 d胚珠中高调表达;Ghir_A02G015550、Ghir_A04G014830和Ghir_D13G009230在 NDM8的种子中高调表达,Ghir_A13G010390和Ghir_D02G015800在NDM8的纤维中高调表达。根据基因注释信息和KEGG比对结果,Ghir_A02G015550参与植物激素信号转导通路,Ghir_A04G014830参与 TCA循环和次生代谢物质生物合成通路,Ghir_D02G015800参与光合生物的碳固定、次生代谢物质生物合成和氨基酸生物合成通路,Ghir_D10G018940参与次生代谢物质生物合成通路,Ghir_D13G005390参与次生代谢物质生物合成、氨基酸生物合成和苯丙氨酸、酪氨酸和色氨酸生物合成通路,Ghir_D13G009230参与次生代谢物质生物合成通路,Ghir_A13G010390和Ghir_D13G010980参与次生代谢物质生物合成、环丙烷生物合成和氰基氨基酸代谢通路(表6)。
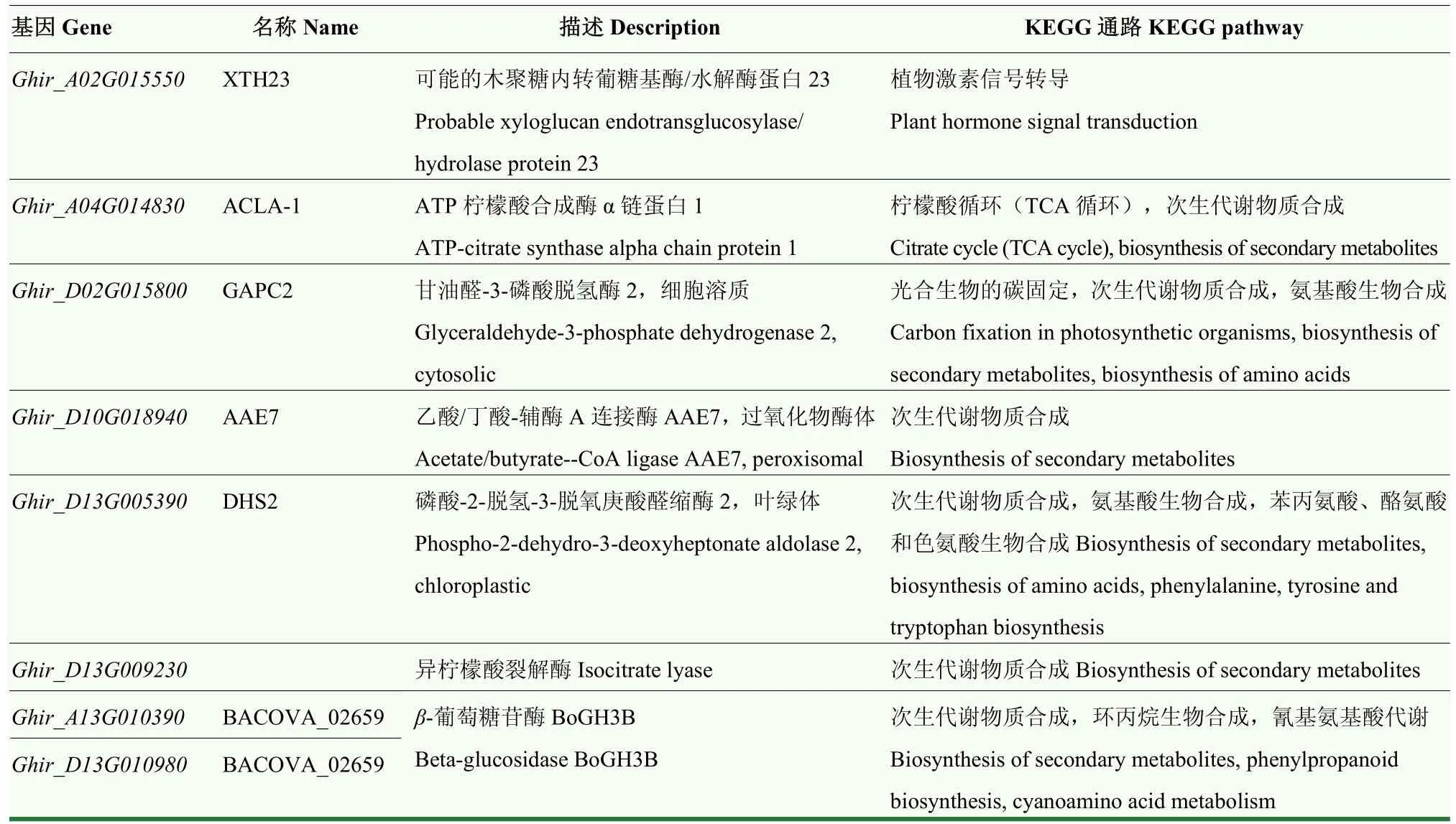
表6 在不同组织中高调表达的8个基因信息Table 6 Information of the 8 genes highly expressed in different tissues

图2 前20个GO富集条目Fig.2 The top 20 items of GO enrichment
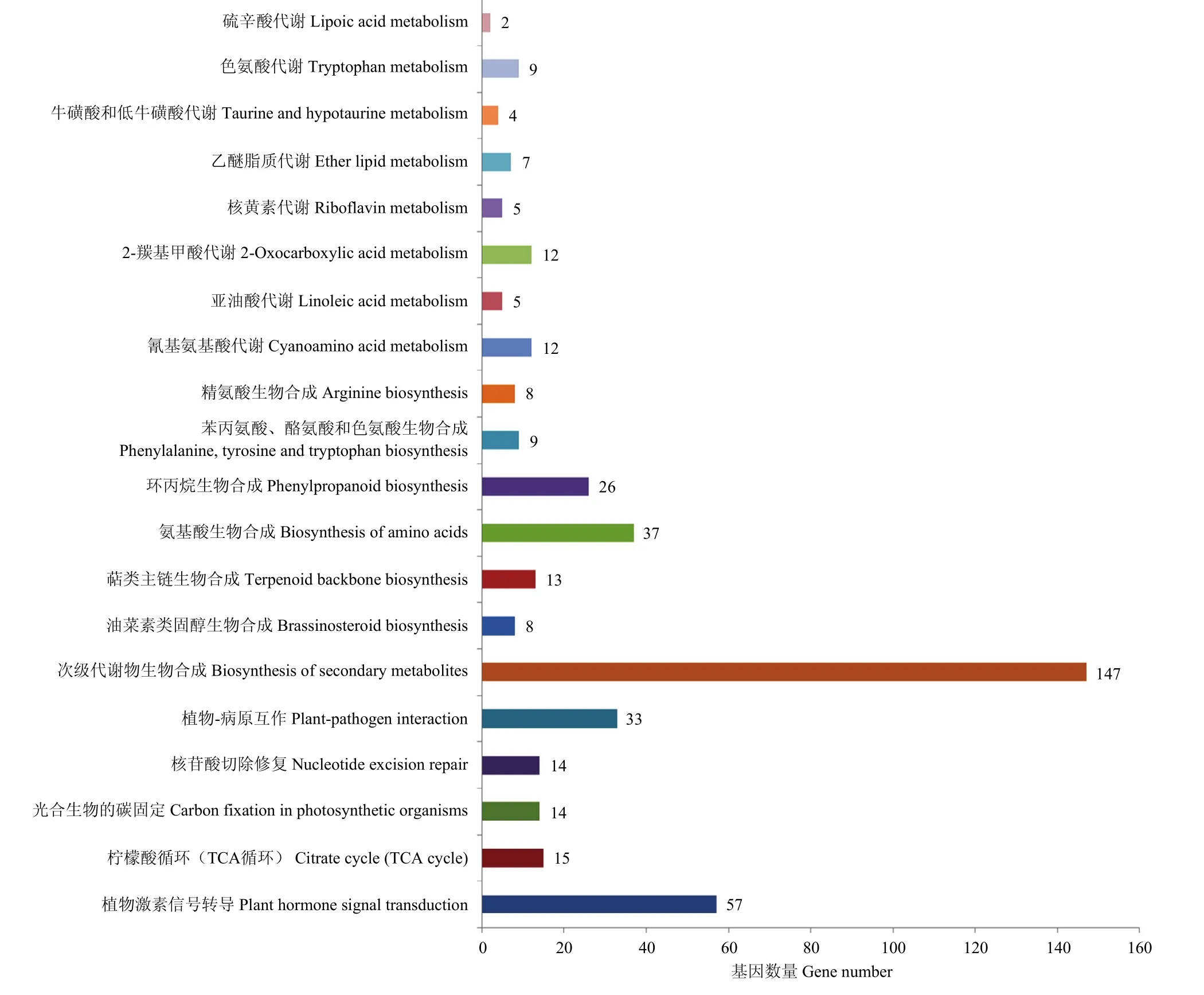
图3 前20个KEGG通路Fig.3 The top 20 KEGG pathways
3 讨论
3.1 利用GBS技术构建了一张高质量的SNP遗传图谱
以SSR等标记构建遗传图谱,由于标记的数量较少、多态性较差等原因,图谱的标记密度较低。如LI等[25]构建了4个F2群体的SSR遗传图谱,但是标记间平均距离最小的为8.25 cM,JAMSHED等[26]构建了一个RIL群体的SSR遗传图谱,标记间的平均距离为5.2 cM。利用高通量测序技术开发SNP标记可显著提高图谱的标记密度,有助于获得稳定性好、精确度高的QTL,并且可进行候选基因分析。例如ZHANG等[2]利用SLAF-seq、芯片以及SSR标记等构建了一张包含8 295个标记、总图距5 197.17 cM的高密度遗传图谱,在17个环境中定位到198个稳定QTL和37个QTL簇,利用RNA-seq数据分析了关键QTL簇内参与调控棉花产量和纤维品质的候选基因,显著提高了基因挖掘的效率。SI等[27]利用RAD-seq构建了一张包含6 303个bin、总图距为5 057.13 cM的海陆种间遗传图谱,并且在 15个位点内发现了存在非同义突变SNP的基因,分别预测了1个产量和1个光合效率的候选基因。因此,说明高通量测序是进行高效QTL定位和候选基因分析的关键技术。
GBS是一种效率高、成本低、广泛应用的简化基因组测序技术[18-19,28]。棉花中,LI等[20]以F2群体为材料,利用GBS技术构建了一张包含3 978个SNP、总图距为2 480 cM的高密度遗传图谱,标记间的平均距离仅有0.62 cM,对棉花的早熟性进行QTL定位和基因注释,根据基因表达量,在稳定的QTL位点内筛选到2个调控早熟性的候选基因。DIOUF等[16]以F2:3群体为材料,构建了一张包含5 178个SNP、总图距为4 768.1 cM的遗传图谱,通过对纤维产量和品质进行QTL定位和基因注释,筛选到5个候选基因。由此说明,GBS技术在棉花中同样具有高效的应用价值。本研究利用GBS技术,在F2群体中开发了1 305 642个SNP标记,构建了一张包含16 088个SNP的遗传图谱,标记间的平均距离仅有0.27 cM,并通过共线性分析证明了图谱质量。在标记数量和密度方面,本研究所构建的遗传图谱与JIA等[29](6 295个SNP、4 071.98 CM)、LI等[20](3 978个SNP、2 480 cM)、ZHANG等[15](5 521个SNP、3 259.378 cM)、GU等[17](6 187个bin、4 478.98 cM)、DIOUF等[16](5 178个SNP、4 768.1 cM)等利用高通量测序技术构建的遗传图谱具有一定可比性。
3.2 产量QTL和候选基因分析
产量的形成是光合产物积累的过程,棉花的产量主要有棉籽和棉纤维两部分。油分和蛋白质是棉籽的主要成分,二者的含量分别为13.6%—24.7%和12.0%—23.0%[30-31],是重要的植物性饲料原料。棉纤维的主要成分为纤维素(~95%)[32-33],是纺织业及其他工业最重要的天然纤维原料。本研究通过构建高密度SNP遗传图谱,在3个群体中定位到108个QTL,其中,稳定QTL共有16个,主效QTL共有10个。有4个稳定QTL的增效基因在不同世代群体中的来源不同,可能是数量性状受环境的影响显著,或者这4个位点存在上位性效应,导致等位基因在不同环境下表达了不同的效应。通过与前人研究结果比较,30个 QTL位点与已公布棉花产量QTL位置重合或接近(电子附表1),其余78个QTL在所比对的文献中未见报道。可能是由于本研究所用的冀丰1271和冀丰173均为自育品种,且首次用于遗传图谱的构建和产量相关性状的QTL定位研究,因此发现了较多新的QTL位点。
在21个主效和稳定的QTL内共注释到3 415个基因,通过KEGG比对分析,有279个基因参与信号转导、物质代谢与合成等通路,主要涉及脂质、亚油酸、氨基酸、次生代谢物质的合成与代谢以及植物激素信号转导、碳固定、TCA循环等能量代谢途径。通过分析基因在TM-1和NDM8不同组织中的表达模式发现,Ghir_D02G015800、Ghir_A13G010390和Ghir_D13G010980在TM-1的胚珠和纤维中均高调表达,Ghir_D10G018940和Ghir_D13G005390在TM-1的 0和 1 d胚珠中高调表达,Ghir_A02G015550、Ghir_A04G014830和Ghir_D13G009230在NDM8的种子中高调表达,Ghir_A13G010390和Ghir_D02G015800在NDM8的纤维中高调表达。其中,Ghir_A02G015550参与植物激素信号转导通路,Ghir_A04G014830参与TCA循环和次生代谢物质生物合成通路,Ghir_D02G015800参与光合生物的碳固定、次生代谢物质生物合成和氨基酸生物合成通路,Ghir_D10G018940参与次生代谢物质生物合成通路,Ghir_D13G005390参与次生代谢物质生物合成、氨基酸生物合成和苯丙氨酸、酪氨酸和色氨酸生物合成通路,Ghir_D13G009230参与次生代谢物质生物合成通路,Ghir_A13G010390和Ghir_D13G010980参与次生代谢物质生物合成、环丙烷生物合成和氰基氨基酸代谢通路。这些通路均涉及物质的合成与代谢,可能是通过调控纤维和种子的发育,影响衣分和子指,进而影响棉花的产量,是重要的候选基因。
4 结论
构建了一张包含16 088个SNP、总图距为4 282.81 cM的高密度遗传图谱,定位到108个产量相关QTL,有 5个主效 QTL可在多个群体中定位到,鉴定到 8个基因在纤维、胚珠或种子中高表达,可能为调控棉花产量形成的重要候选基因。
猜你喜欢
中国棉花加工(2023年4期)2023-09-27 11:28:58
今日农业(2021年11期)2021-08-13 08:53:24
少先队活动(2020年12期)2021-01-14 01:47:40
中国纤检(2019年5期)2019-12-09 16:06:20
中国棉花(2018年7期)2018-08-29 00:54:08
中成药(2017年3期)2017-05-17 06:09:01
领导科学论坛(2016年9期)2016-06-05 14:59:58
安徽农学通报(2016年7期)2016-05-03 02:11:14
遗传(2014年3期)2014-02-28 20:58:49
世界科学(2014年8期)2014-02-28 14:58:31
