
微核糖核酸-125a通过调控细胞凋亡和炎症反应延缓阿尔茨海默病进展的研究
2023-03-07 00:40:10周冰凌吴家顺李俐娟
上海医学 2023年1期
周冰凌 邵 卫 邱 昕 吴家顺 徐 雷 李俐娟
阿尔茨海默病(Alzheimer’s disease,AD)是一种诱因不明的神经退行性疾病,其病理学特点为β-淀粉样蛋白(amyloid β-protein,Aβ)沉积造成的神经炎斑、神经元纤维缠结、神经元死亡,其主要临床表现为记忆能力、认知判断能力、语言理解能力等功能的进行性衰退[1]。我国流行病学资料显示,AD患病率日益增高,成为近年来威胁老年人群健康及生活质量的重要疾病之一[2]。目前,有效预防和治疗AD的手段较为有限。有研究[3-4]显示,促炎因子水平与神经胶质细胞损伤程度呈正相关,且系统性炎症能够催生神经毒素,进而促进AD的发生。因此,抗炎药物对于延缓AD的发生、发展有一定的作用。微核糖核酸-125a(mircoRNA-125a,简称miR-125a)表达与脑源性神经营养因子(brain-derived neurotrophic factor,BDNF)水平呈正相关,可调控神经元的增殖和凋亡,并参与帕金森病、自身免疫性脑脊髓炎等神经退行性疾病的发生[5-7]。此外,miR-125a通过磷脂酰肌醇-3-激酶/蛋白激酶B/雷帕霉素靶蛋白(PI3K/Akt/mTOR)相关信号通路调控炎症反应[8-9]。基于上述证据,本研究团队假设miR-125a在AD的发生、发展中起着重要作用,然而目前鲜见相关报道。本研究拟采用Aβ1-42毒化神经生长因子(nerve growth factor, NGF)诱导的PC12细胞株建立PC12 AD模型细胞,旨在探究miR-125a对PC12 AD模型细胞的凋亡、总神经突生长长度和炎症因子水平的影响。
1 材料和方法
1.1 细胞培养 PC12细胞株购自中国典型培养物保藏中心。在37 ℃、体积分数为0.05的CO2且饱和湿度条件下,以含5%胎牛血清(美国赛默飞世尔科技有限公司)和10%灭活马血清(美国赛默飞世尔科技有限公司)的RPMI-1640培养基(美国Sigma-Aldrich公司)培养 PC12细胞株。
1.2 PC12 AD模型细胞构建 参考文献[10]的方法,建立PC12 AD模型细胞。首先,将PC12细胞株接种于24孔板,密度为1×105/mL。加入含20 ng/mL NGF(美国Sigma-Aldrich公司)、10%胎牛血清的RPMI-1640培养基培养,在37 ℃、体积分数为0.05的CO2条件下诱导细胞分化72 h。随后,将已在37 ℃预先孵育(目的为促进多肽聚合)7 d的Aβ1-42(美国Sigma-Aldrich公司)溶解于二甲基亚砜(DMSO)中配制成1 mmol/L的溶液,储存于-20 ℃待用。取1 μmol/L低聚Aβ1-42加入NGF诱导分化后的PC12细胞株中培养24 h以构建PC12 AD模型细胞,作为Aβ1-42组;将NGF诱导分化后未经Aβ1-42处理的PC12细胞株,作为对照组。
向上述两组各孔中加入CCK-8[东仁化学科技(上海)有限公司]试剂10 μL, 37 ℃孵育2 h,应用酶标仪(美国Bio-Tek公司)检测波长为450 nm处的吸光度计算细胞活性,以百分数表示。采用实时荧光定量聚合酶链式反应(RT-qPCR)检测对照组和Aβ1-42组中的miR-125a相对表达量。
1.3 转染 miR-125a 模拟物(mimic)、对照mimic、miR-125a抑制剂(inhibitor)和对照inhibitor均由广州市锐博生物科技有限公司合成。将PC12 AD模型细胞接种于24孔板中,密度为1×105/mL,使用Lipofectamine 2000(美国Invitrogen公司)将miR-125a mimic、对照mimic、miR-125a inhibitor及对照inhibitor分别转染至PC12 AD模型细胞中,并在37 ℃、体积分数为0.05的CO2条件下孵育24 h后,采用RT-qPCR检测各组细胞中miR-125a相对表达量。孵育48 h后,在显微镜下观察、计算总神经突生长长度;使用Hoechst/碘化丙啶(propidium iodide,PI;美国Sigma-Aldrich公司)试剂检测细胞凋亡情况;同时,收集细胞培养上清液,按照ELISA试剂盒(美国R&D Systems公司)操作说明书测定炎症因子(TNF-α、IL-1β、IL-6和IL-17)水平。
1.4 RT-qPCR 使用TRIzolTMReagent(美国赛默飞世尔科技有限公司)提取总RNA,并使用iScriptTMcDNA Synthesis Kit逆转录试剂盒(美国Bio-Rad公司)将提取的RNA反转录为cDNA。以反转录产物为模板,使用QuantiNova SYBR Green PCR试剂盒(德国凯杰公司)进行RT-qPCR。以U6作为内参,采用2-△△Ct方法计算miR-125a相对表达量;以β-actin作为内参,采用2-△△Ct方法计算BDNF相对表达量。引物序列:miR-125a正向序列为5’-ACA CTC CAG CTG GGT CCC TGA GAC CCT TTA AC-3’;反向序列为5’-TGT CGT GGA GTC GGC AAT TC-3’。U6正向引物为5’-CGC TTC GGC AGC ACA TAT ACT A-3’;反向序列为5’-ATG GAA CGC TTC ACG AAT TTG C-3’。BDNF正向序列为5’-GAC GGT CAC AGT CCT GGA GAA-3’;反向序列为5’-GCA GCC TTC CTT CGT GTA ACC-3’。β-actin正向序列为5’-CTA TCG GCA ATG AGC GGT TCC-3’;反向序列为5’-GCA CTG TGT TGG CAT AGA GGT C-3’。
1.5 细胞凋亡检测 转染48 h后,向PC12 AD模型细胞中加入8 mol/L Hoechst 33342 和1.5 mol/LPI在37 ℃下孵育0.5 h后,应用倒置荧光显微镜(德国Leica公司)摄取细胞图片。在200倍视野下,应用Image Pro Plus软件(美国Media Cybernetics公司)统计Hoechst 33342阳性和PI阳性细胞数量,计算细胞凋亡率。
1.6 神经突生长测量 转染48 h后,于显微镜(德国Leica公司)下观察细胞形态,并应用Imaging Software Presage软件(美国Advanced Imaging Concepts公司)测量神经突生长长度,以所有细胞神经突生长长度之和除以细胞总数,计算每个细胞总神经突生长长度。
1.7 乳酸脱氢酶(lactate dehydrogenase,LDH)细胞毒性检测 转染48 h后,收集2组100 μL细胞培养基置于96孔板内,并加入100 μL 预配置的LDH工作液[东仁化学科技(上海)有限公司]。37 ℃避光孵育30 min后,加入50 μL反应终止液,应用酶标仪检测波长为490 nm处的样本吸光度。在空白组中加入20 μL Lysis缓冲液后[东仁化学科技(上海)有限公司] ,于37 ℃孵育30 min,收集100 μL上清液用于LDH毒性检测。细胞毒性= [(Aβ1-42组吸光度-空白组吸光度)÷(对照组吸光度-空白组吸光度)]×100%。
1.8 免疫印迹实验 转染48 h后,收集细胞株并应用RIPA lysis缓冲液(美国Sigma-Aldrich公司)在冰上对细胞进行裂解。收集上清液,使用BCA 蛋白定量试剂盒 (上海碧云天生物技术有限公司)对细胞蛋白质进行定量。取10 μg总蛋白质,经热变性后,以4%~20%预制胶(上海碧云天生物技术有限公司)对蛋白质进行十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDS-PAGE),后将目的蛋白质转移至硝酸纤维素膜(美国Sigma-Aldrich公司)。在37 ℃下,硝酸纤维素膜经5%小牛血清(BSA,上海碧云天生物技术有限公司)封闭90 min后,在4 ℃,与一抗抗体共孵育过夜。收集一抗抗体后,滴加稀释后的二抗抗体至硝酸纤维素膜,于37 ℃孵育1 h。洗去二抗抗体后,滴加增强化学发光法的发光试剂(上海碧云天生物技术有限公司)至硝酸纤维膜对目的蛋白质进行显影。应用Image J 1.8软件对蛋白质条带进行定量。该实验中所用抗体为BDNF一抗抗体(1∶1 000, 江苏亲科生物研究中心有限公司)、β-actin一抗抗体(1∶10 000,江苏亲科生物研究中心有限公司)、辣根过氧化物酶(HRP)标记的山羊抗兔抗体(1∶10 000,江苏亲科生物研究中心有限公司)。
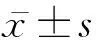
2 结 果
2.1 两组细胞活性和miR-125相对表达量的比较 Aβ1-42组细胞活性为(35.60±4.38)%,较对照组的(100.30±4.51)%显著降低(P<0.001),表明成功构建PC12 AD模型细胞。Aβ1-42组miR-125a相对表达量为0.86±0.04,显著少于对照组的0.99±0.03(P<0.05)。
2.2 两组细胞转染后miR-125a相对表达量的比较 miR-125a mimic组miR-125a相对表达量为3.95±0.24,较对照mimic组的1.00±0.13显著增加(P<0.001)。miR-125a inhibitor组miR-125a相对表达量为0.31±0.08,较对照inhibitor组的0.99±0.10显著减少(P<0.001),表明转染成功。
2.3 miR-125a对PC12 AD模型细胞凋亡的影响 miR-125a mimic组细胞凋亡率为(7.55±1.56)%,显著低于对照mimic组的(13.00±1.11)%(P<0.05);miR-125a inhibitor组细胞凋亡率为(19.34±2.41)%,显著高于对照inhibitor组的(13.11±2.04)%(P<0.01)。miR-125a mimic组LDH毒性细胞比例为(6.14±1.20)%,显著低于对照mimic组的(15.43±2.55)%(P<0.05);miR-125a inhibitor组LDH毒性细胞比例为(23.07±3.86)%,显著高于对照inhibitor组的(14.79±2.26)%(P<0.05)。
2.4 miR-125a对PC12 AD模型细胞总神经突生长长度的影响 miR-125a mimic组总神经突生长长度为(34.54±2.95) μm,较对照mimic组的(23.93±2.64) μm显著增长(P<0.01)。miR-125a inhibitor组总神经突生长长度为(18.47±2.27) μm, 较对照inhibitor组的(24.62±1.52) μm显著缩短(P<0.05)。见图1。

A miR-125a mimic组 B 对照mimic组 C miR-125a inhibitor组 D 对照inhibitor组图1 荧光显微镜下观察miR-125a对PC12 AD模型细胞总神经突生长长度的影响(×40)
2.5 miR-125a对PC12 AD模型细胞炎症因子水平的影响 miR-125a mimic组中TNF-α、IL-1β、IL-6、IL-17水平分别为(22.72±4.06)、(18.32±3.50)、(12.52±1.47)和(21.12±1.81) pg/mL,相较对照mimic组的(42.48±2.59)、(38.01±3.29)、(24.81±3.82)和(44.38±4.15) pg/mL均显著降低(P<0.01或0.001)。miR-125a inhibitor组中TNF-α、IL-1β、IL-6、IL-17水平分别为(66.81±5.52)、(59.78±7.15)、(50.53±2.75)和(63.10±4.95) pg/mL,相较对照inhibitor组的(40.28±4.36)、(37.24±5.92)、(24.54±5.13)和(43.06±3.75) pg/mL均显著增高(P<0.05或0.01)。
2.6 miR-125a对PC12 AD模型细胞BDNF水平的影响 miR-125a mimic组BDNF mRNA和蛋白质相对表达量分别为1.69±0.25、1.73±0.30, 均较对照mimic组的1.00±0.18、0.94±0.11显著增加(P值均<0.01)。miR-125a inhibitor组BDNF mRNA和蛋白质相对表达量分别为0.43±0.14、0.25±0.14,均较对照inhibitor组的1.08±0.13、0.96±0.13显著减少(P值均<0.01)。见图2。
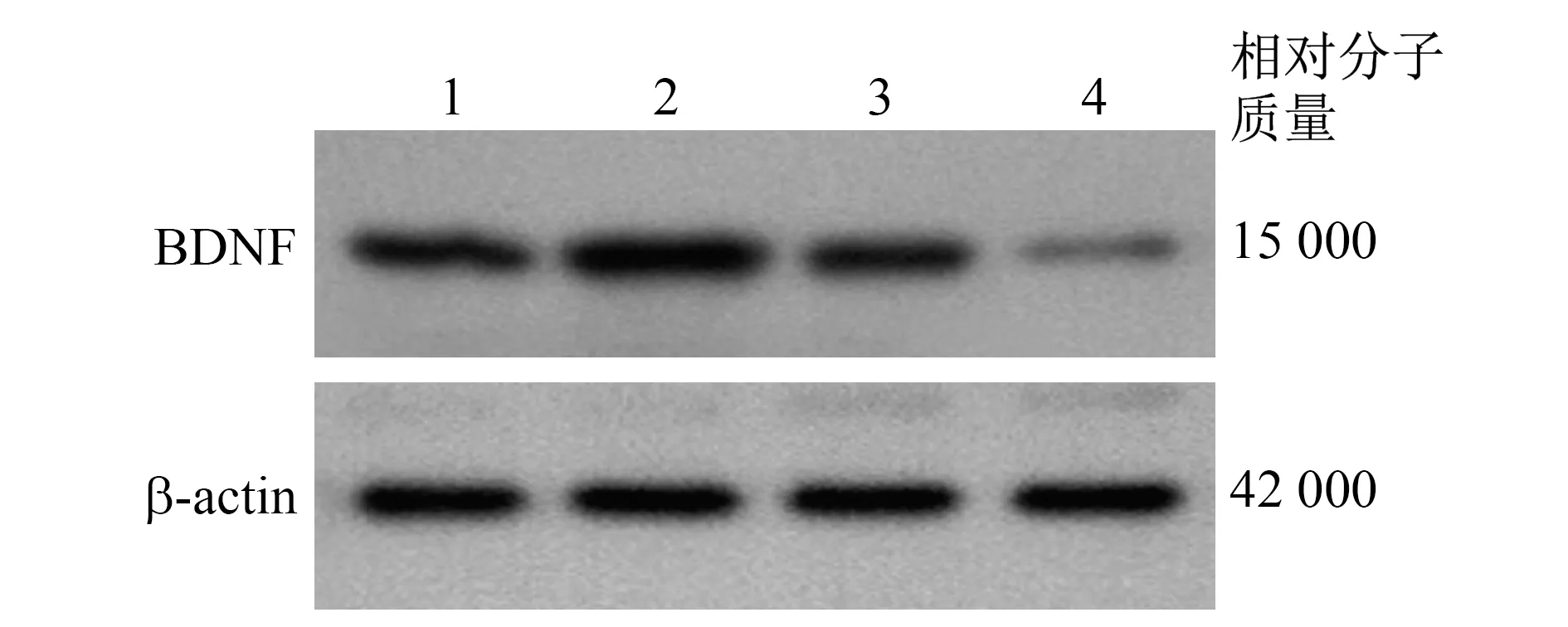
1 对照mimic组 2 miR-125a mimic组 3 对照inhibitor组 4 miR-125a inhibitor组图2 miR-125a对PC12 AD模型细胞BDNF蛋白质表达的影响
3 讨 论
本研究通过使用低聚Aβ1-42加入NGF诱导分化后的PC12细胞株构建PC12 AD模型细胞,发现在PC12 AD模型细胞中:①miR-125a表达水平较对照组更低;②miR-125a抑制细胞凋亡,并促进细胞总神经突长度的增长;③miR-125a降低细胞炎症因子水平。
AD是老年人群常见的疾病之一,据WHO不完全统计数据显示,全球65岁以上人群AD的患病率约4%~7%,且患病率随着年龄增长而日益增高,85岁以上人群AD的患病率可高达30%[11]。AD典型的早期临床特征包括学习、记忆能力损伤,随之是注意力、行为能力、语言功能、空间视觉功能、感知力、社交能力的丧失[3]。此外,AD会造成患者逐渐失去日常生活自理能力,不仅给患者及其家庭带来了沉重的精神压力和经济负担,对整个社会医疗资源都是巨大的挑战[12]。目前,针对AD的具体发病机制存在多种假说,常见的包括胆碱能神经元假说、Aβ毒性假说和Tau蛋白假说等。近年来,在早期研究的基础上,炎症假说越来越受到关注[3]。因此,亟须探究炎症在AD发病机制或参与AD发生、发展过程中的生物标志物。
研究[5-7, 13]结果表明,miR-125a可影响神经元细胞的增殖与凋亡,并参与一些神经退行性疾病的病理过程。例如,miR-125a表达与BDNF蛋白质水平呈正相关,可通过调控BDNF蛋白质的表达影响神经元细胞的增殖与凋亡[5]。此外,miR-125a在自身免疫性脑脊髓炎的脊髓前角细胞中异常表达,通过调控维生素D受体活动,参与脑脊髓炎的发生[7]。近期的研究[6]显示,帕金森病患者的脑脊液中miR-125a水平较健康人群显著降低,miR-125a可作为早期诊断帕金森病的生物标志物。此外,miR-125a在炎症反应中的作用也有所报道。炎性肠病的机制研究[14]发现,miR-125a通过靶向E-二十六转录因子1(E-twenty six transcription factor-1,ETS-1)蛋白,从而抑制IFN-γ、TNF-α和IL-17A等促炎因子的分泌,进而减少对肠黏膜的炎症损伤。亦有研究[15]结果表明,miR-125a能够减少巨噬细胞M1表型的表达,同时促进M2表型的表达,以调控巨噬细胞极化作用并激活巨噬细胞,参与脓毒症、肺炎等相关疾病的病理过程,发挥抑制炎症的作用。关于甲状腺炎的研究[16]结果显示,甲状腺炎患者血清中miR-125a异常表达;进一步的机制研究结果表明,miR-125a通过调控PI3K/Akt/mTOR信号通路,抑制促炎因子的表达,并影响甲状腺炎模型细胞的增殖与凋亡,参与细胞自噬。基于miR-125a在神经退行性疾病及炎症中的重要调控作用,本研究假设miR-125a能够影响AD细胞凋亡、总神经突生长长度和炎症因子水平,从而参与AD的病理过程。结果发现,在通过低聚Aβ1-42诱导神经损伤的PC12 AD模型细胞中,miR-125a相对表达量较对照组更低。可能是由于低聚Aβ1-42促进神经炎症,提高了总体炎症水平,进而降低了miR-125a这一抑炎因素的水平,然而这一推论需要进一步的研究支持。
为了进一步探究miR-125a是否对PC12 AD模型细胞的细胞活性有调控作用,本课题组将miR-125a mimic、对照mimic、miR-125a inhibitor及对照inhibitor分别转染至PC12 AD模型细胞,对各组细胞的总神经突生长长度和凋亡进行了检测。结果显示,在PC12 AD模型细胞中,miR-125a促进总神经突长度的增长,并抑制细胞凋亡。可能的原因包括:①在PC12 AD模型细胞中,miR-125a通过调控PI3K/Akt/mTOR信号通路,抑制炎症的发生,进而延缓AD的进展,促进总神经突的生长,抑制细胞凋亡。②在PC12 AD模型细胞中,miR-125a可能通过诱导巨噬细胞向M2型极化,增强M2表型标记物(包括CD163、CD209等)的基因表达,抑制促炎因子的形成,增加抗炎因子的分泌,从而形成一种抗炎的微环境,修复细胞损伤,继而抑制细胞凋亡。同时,本课题组通过比较miR-125a mimic组与对照mimic组、miR-125a inhibitor组与对照inhibitor组的TNF-α、IL-1β、IL-6、IL-17水平,发现在PC12 AD模型细胞中,miR-125a可调控降低炎症因子水平。这一结果提示miR-125a可能通过降低炎症因子水平延缓AD的发生、发展。
综上所述,miR-125a可能对Aβ1-42毒化的分化PC12细胞株具有保护作用,其有望为AD的预防和诊断提供新的思路,并为AD的治疗提供新的靶点。
猜你喜欢
数学小灵通(1-2年级)(2020年9期)2020-10-27 03:24:46
中成药(2017年9期)2017-12-19 13:34:27
作文大王·低年级(2017年11期)2017-12-05 00:08:45
小学生学习指导(低年级)(2017年12期)2017-11-22 06:22:39
CHINESE JOURNAL OF AERONAUTICS(2017年1期)2017-11-21 12:54:14
中成药(2017年5期)2017-06-13 13:01:12
读写算(上)(2015年6期)2015-11-07 07:17:55
医学研究杂志(2015年11期)2015-06-10 06:44:03
中国当代医药(2015年16期)2015-03-01 02:03:11
中国医药导报(2015年27期)2015-02-28 22:08:02
